Zinc Finger in Every Pie? Protective Protein Partners with Many ALS Culprits
Quick Links
Repeats of the seemingly innocuous hexanucleotide sequence GGGGCC cause amyotrophic lateral sclerosis (ALS) more than any other known factor. Taking one step closer to understanding how, researchers led by Brian Black at the University of California, San Francisco, have uncovered a zinc finger protein, Zfp106, which associates with the fateful sequence, as well as with several RNA binding proteins implicated in the disease, including TDP-43 and FUS. As described in eLife on January 10, mice lacking Zfp106 succumb to a progressive, ALS-like disorder, while expressing the zinc finger protein in a C9ORF72 fly model suppressed motor neuron disease. The study adds to other recent reports linking C9ORF72 to the derailment of RNA metabolism and processing. Exactly how Zfp106 dampens the neurotoxicity caused by C9ORF72, and whether the protein’s protective prowess extends to other forms of ALS, are exciting questions that remain unanswered, Black told Alzforum.
Compared to healthy people, who have between two and 23 repeats of the G4C2 sequence in their C9ORF72 gene, people with ALS can have hundreds to thousands. While some researchers contend that aggregates of poly-dipeptides translated from expanded C9ORF72 RNA harms neurons, others claim that the G4C2 repeat RNAs do the damage. To investigate, first author Barbara Celona and colleagues used a string of eight G4C2 repeats to fish for proteins that bind the RNA expansions. The researchers pulled out more than a dozen known and predicted RNA binding proteins. Celona decided to focus further on Zfp106 because the human gene lies in a region on chromosome 15 implicated in a rare recessive form of ALS (see Hentati et al., 1998). Furthermore, Zfp106 knockout mice have a progressive neuromuscular disorder (see Anderson et al., 2016; Joyce et al., 2016).
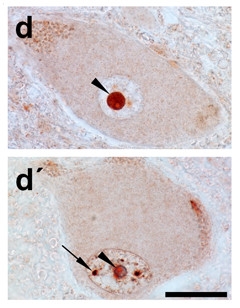
Protein Protector?
In normal motor neurons, Zfp106 (red) resides primarily in the nucleolus (arrowheads), but also in small puncta in the nucleus (arrow). [Courtesy of Celona et al., eLife 2017.]
In a cultured neuronal cell line and in lower motor neurons from postmortem human spinal cord samples from people free of ALS, the researchers found Zfp106 primarily located in the nucleolus and occasionally in clusters inside the nucleus. Using mass spectrometry to identify proteins that buddy up with Zfp106 revealed 39 partners, many being RNA binding proteins. These included TDP-43 and FUS, as well as the nucleolar protein Nucleolin, and the nuclear pore component, Nup107. Together, these findings placed Zfp106 right at the heart of predicted ALS hotspots: the RNA processing, metabolism, and splicing centers of the nucleolus and nucleus.
To investigate the role of Zfp106 in vivo, the researchers characterized Zfp106 knockout mice that were generated as part of a large-scale phenotypic screen called the Sanger Mouse Genetics Project. The Zfp106 knockout animals—in which the β-galactosidase gene replaced part of Zfp106—behaved similarly to wild-type mice for the first month of life, but then all of them started showing signs of muscle weakness and loss. By three months of age, the knockout animals had extensive muscular atrophy and degeneration of the muscle fibers, as well as a widespread death of choline acetyltransferase (ChAT)-positive motor neurons in the spinal cord. A separate Zfp106-deficient mouse line that the researchers generated via CRISPR struggled with the same pathologies. To see if deficiency of Zfp106 specifically in motor neurons drove their neurodegeneration, the researchers next crossed the knockout mice with animals expressing Zfp106 under control of the Mnx1 promoter, which is active only in motor neurons. Offspring were largely spared from cell death, and from the muscle loss and weakness in the Zfp106 knockouts.
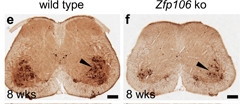
Death without Zpf106.
ChAT-positive motor neurons (dark brown) in spinal cords from Zpf106 knockout mice (right) take a major hit. [Courtesy of Celona et al., eLife 2017.]
How would Zfp106 behave in a C9ORF72 model of ALS? To find out, the researchers turned to flies. In a Drosophila model expressing 30 copies of G4C2, half of the insects die before emerging from their pupal case, and those that do manage to creep out are stricken with severe movement problems. Co-expressing Zfp106 in these insects allowed the embryos to survive past the pupal stage, and partially suppressed the locomotor defects. These findings support the idea that Zfp106 dampens the neurodegenerative effects of C9ORF72 repeat expansions.
Though the function of Zfp106, especially in humans, is unknown, Black proposed that the protein might latch onto C9ORF72 RNA and somehow prevent translation of dipeptide repeats, which he views as the toxic entities in C9-ALS. If so, perhaps therapeutic strategies that bestow motor neurons with extra Zfp106, or small molecules that mimic its binding to RNA, could stave off the disease, he told Alzforum.
Interestingly, two recent studies reported that the dipeptide repeats translated from C9ORF72 expansions associated with some of the same ALS-associated RNA binding proteins that Zfp106 does, and that these partnerships fouled up the function of so-called liquid organelles such as the nucleolus, where RNA metabolism and processing take place (see Oct 2016 news). While Black would not speculate on whether Zfp106 binding to these proteins also affected liquid organelle dynamics, he said that it is certainly possible, given the close quarters the proteins all share in densely packed organelles.
How might Zfp106 play a role in other forms of ALS without C9ORF72 repeat expansions? “This is a major question that needs to be answered next,” commented Abraham Acevedo-Arozena of the Hospital Universitario de Canarias in the Canary Islands, Spain. Acevedo-Arozena, who led one of the studies first characterizing the neuromuscular disorder in Zfp106 knockout mice, added that it is possible the zinc finger protein could protect by associating with RNA binding proteins implicated in the disease, including TDP-43 and FUS, which are known to translocate from the nucleus to the cytoplasm in neurons affected by ALS. Figuring out what the protein does, and whether it can broadly protect against sporadic forms of the disease, are the next major challenges, he added.—Jessica Shugart
References
News Citations
Paper Citations
- Hentati A, Ouahchi K, Pericak-Vance MA, Nijhawan D, Ahmad A, Yang Y, Rimmler J, Hung W, Schlotter B, Ahmed A, Ben Hamida M, Hentati F, Siddique T. Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers. Neurogenetics. 1998 Dec;2(1):55-60. PubMed.
- Anderson DM, Cannavino J, Li H, Anderson KM, Nelson BR, McAnally J, Bezprozvannaya S, Liu Y, Lin W, Liu N, Bassel-Duby R, Olson EN. Severe muscle wasting and denervation in mice lacking the RNA-binding protein ZFP106. Proc Natl Acad Sci U S A. 2016 Aug 2;113(31):E4494-503. Epub 2016 Jul 14 PubMed.
- Joyce PI, Fratta P, Landman AS, Mcgoldrick P, Wackerhage H, Groves M, Busam BS, Galino J, Corrochano S, Beskina OA, Esapa C, Ryder E, Carter S, Stewart M, Codner G, Hilton H, Teboul L, Tucker J, Lionikas A, Estabel J, Ramirez-Solis R, White JK, Brandner S, Plagnol V, Bennet DL, Abramov AY, Greensmith L, Fisher EM, Acevedo-Arozena A. Deficiency of the zinc finger protein ZFP106 causes motor and sensory neurodegeneration. Hum Mol Genet. 2016 Jan 15;25(2):291-307. Epub 2015 Nov 24 PubMed.
Further Reading
Primary Papers
- Celona B, Dollen JV, Vatsavayai SC, Kashima R, Johnson JR, Tang AA, Hata A, Miller BL, Huang EJ, Krogan NJ, Seeley WW, Black BL. Suppression of C9orf72 RNA repeat-induced neurotoxicity by the ALS-associated RNA-binding protein Zfp106. Elife. 2017 Jan 10;6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Sheffield
Celona et al. describe characterization of a potential therapeutic target for ALS patients with GGGGCC-repeat expansions of the C9ORF72 gene, and potentially ALS more broadly. The work centers on the protein Zfp106—a zinc finger protein with unknown function. The authors describe how overexpression of Zfp106 can rescue toxicity in a Drosophila model of C9ORF72-ALS expressing 30 tandem GGGGCC-repeats.
One of the most interesting aspects of this work is the suggestion that Zfp106 might be relevant more broadly in ALS. The authors point to the fact that the Zfp106 gene lies within a region strongly linked to a rare recessive form of ALS (Hentati et al., 1998). Moreover they have used co-immunoprecipitation and mass spectroscopy to identify Zfp106 binding partners, which include proteins linked to ALS such as TDP-43. TDP-43 is itself mutated in rare cases of ALS but more interestingly, it forms a prominent component of motor neuron cytoplasmic inclusions in more than 98 percent of ALS cases. The suggestion is that loss of Zfp106 function might be part of the final common pathway of neurotoxicity in ALS. To add weight to this hypothesis, the authors have characterised Zfp106-null mice and described an ALS-like phenotype including motor neuron degeneration.
The authors have speculated on, but not demonstrated, a mechanism for the proposed role of Zfp106 in neurotoxicity. They show that the Zfp106 protein binds specifically to GGGGCC-repeat RNA and they propose that, as has been proposed for other RNA-binding proteins, sequestration by the GGGGCC-repeat RNA could lead to a partial loss of Zfp106 function. This leads to a neat hypothesis that loss of Zfp106 function contributes to ALS-neurotoxicity, perhaps via an interaction with TDP-43, and that the loss of Zfp106 function may be caused through sequestration by GGGGCC-repeat RNA in C9ORF72-ALS, or through alternative mechanisms in other forms of ALS. This may be testable. No heterozygous mutant-Zfp106 ALS cases have been identified, which suggests that partial loss of Zfp106 may not be sufficient on its own to cause ALS. However if Zfp106 function is a significant disease modifier, then Zfp106 mutations in combination with other ALS-causing mutations might cause synergistic toxicity. In that case, we might expect to see reported oligogenic cases of ALS with mutations in Zfp106 and another ALS-linked gene. Overexpression of Zfp106 would then potentially become a very attractive and broadly applicable therapeutic target.
References:
Hentati A, Ouahchi K, Pericak-Vance MA, Nijhawan D, Ahmad A, Yang Y, Rimmler J, Hung W, Schlotter B, Ahmed A, Ben Hamida M, Hentati F, Siddique T. Linkage of a commoner form of recessive amyotrophic lateral sclerosis to chromosome 15q15-q22 markers. Neurogenetics. 1998 Dec;2(1):55-60. PubMed.
Make a Comment
To make a comment you must login or register.