A Year in Research—Top 10 Trends of 2014
Quick Links
Are you good for more exercise and losing five pounds, but forgot all about making New Year’s resolutions for your science? Ponder the past year's advances, setbacks, and surprises in AD research, and use them as fodder for new ideas in 2015.
Trials, Therapies, and Diagnostics
Depending on one’s perspective, 2014 was a glass half-empty or half-full. For those expecting approval of a new drug among the various Aβ antibodies in the running, the year started out on a low when in January, papers in the New England Journal of Medicine made it official that Phase 3 trials of bapineuzumab and solanezumab had missed their primary goals. These papers renewed debate that lasted throughout the year and flared up loudly at the Clinical Trials on Alzheimer’s Disease (CTAD) conference in November. Some diagnosed a body blow for the amyloid hypothesis; others spotted in the wreckage some encouraging signals that treatment in the mildest patients engaged biomarkers, improved cognition, and slowed disease. Bapineuzumab is on the scrap heap, but solanezumab is being tested in another Phase 3 trial of mild AD, as well as in a prevention trial by the Dominantly Inherited Alzheimer Network of people carrying early onset mutations in APP and presenilin.
July brought word that Phase 2 trials of crenezumab had similarly fallen short of primary endpoints of cognition and function. Much like solanezumab, this antibody, too, gave faint signs of life with a cognitive benefit at the higher dose in the mildest patients (see Jul 2014 conference story). The crenezumab program emphasized how biomarker analysis in AD trials still requires much refinement. For example, amyloid PET showed no difference between placebo and treatment groups when AD regions were compared to cerebellum, but did begin to show a separation when compared with white matter (see Dec 2014 conference story). Crenezumab is being evaluated in an Alzheimer Prevention Initiative (API) trial in families carrying an autosomal-dominant presenilin mutation.
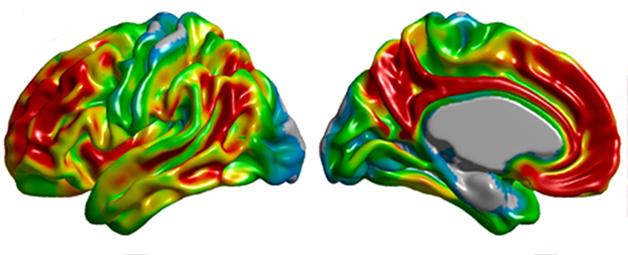
Spatial distribution of amyloid PET tracer binding in Alzheimer's disease. [Image courtesy of Felix Carbonell, Biospective Inc.]
The year ended on another bust, when Roche announced just before Christmas that it was ending an ongoing Phase 2/3 trial of gantenerumab in prodromal AD because it failed to meet the bar in a pre-specified futility analysis (see Dec 2014 news story). The gantenerumab arm of the DIAN-TU prevention trial will continue.
Other immune-based treatments are still in the running. For example, Novartis' CAD106, an active immunotherapy, completed Phase 2 (see Aug 2014 conference story) and was chosen for an upcoming API secondary prevention trial of aged ApoE4 carriers (see Jul 2014 conference story). For its part, Biogen decided to push its antibody aducanumab straight from Phase1b to Phase 3 (see Dec 2014 conference story), and other antibody trials are ongoing.
The failures thus far prompted extensive debate among scientists. Some felt the data showed that Aβ antibodies simply do not work. Other scientists cited specifics of antibody-Aβ binding characteristics, effector function, brain exposure, dosing, target engagement, and disease stage as the reasons antibodies have not worked yet. They insist the field is learning from each trial and immunotherapy evaluation should continue for the time being.
Bioavailability continues to be a problem for immunotherapy on AD, because only trace amounts of injected antibodies make it into the brain. The solution? Hitch the antibody to a protein that freely crosses the blood-brain barrier. Last January, two groups reported progress in delivering anti-Aβ or anti-BACE antibodies into mouse brain by way of transferrin, and November brought news that the system worked in primates (see Jan 2014 news story; Jan 2014 news story; Nov 2014 news story).
On the small-molecule front, BACE inhibitors have become the next big thing. Merck's MK-8931 is furthest along, being tested in two Phase 3 trials, one in prodromal and one in mild to moderate AD. Lilly teamed up with AstraZeneca to run a Phase 2/3 trial of the BACE inhibitor AZD3293 (see Oct 2014 news story) and, at CTAD in November, presented Phase 1 data suggesting strong target engagement and Aβ-lowering in AD patients. Vitae Pharmaceuticals announced top-line data from a single-dose Phase 1 study of its BACE inhibitor, VTP-37948, which lowered CSF Aβ by up to 80 percent (see Oct 2014 news story). Both the API ApoE and DIAN prevention trials will use a BACE inhibitor, but which one remains unclear. Novartis presented preliminary data on NB-360 at the AAIC meeting in July (see Jul 2014 conference story; Aug 2014 conference story).
All the while, academic researchers have continued to urge caution in blocking BACE while potential side effects are still being studied, lest a similar fate meet these inhibitors as befell their γ-secretase counterparts (see Jun 2014 news story; Nov 2014 news story). The year 2014 saw γ-secretase inhibitors slide into oblivion. They had been considered dead as a doornail since investigators stopped the semagacestat Phase 3 in 2013 because of safety concerns; however, a nagging regret expressed largely in private discussion found publication in November 2014, when Bart de Strooper, KU Leuven, Belgium, analyzed why that trial failed. He urged the field to learn much more about the pharmacokinetics of secretase inhibition, and use the semagacestat experience to fill knowledge gaps about γ-secretase as a target rather than simply abandoning it and rushing toward a new, just-as-poorly understood target in BACE1 (De Strooper, 2014).
Some good news came from studies of drugs aimed at calming agitation in AD patients. Both AVP-923, aka Nuedexta, and citalopram showed some promise (see Feb 2014 news story; Oct 2014 conference story). Nuedexta is a mixture of dextromethorphan hydrobromide, the active ingredient in the cough syrup Robitussin, and quinidine sulfate, which prevents the liver enzyme cytochrome P450 from breaking down dextromethorphan. Citalopram, a selective serotonin reuptake inhibitor, carries a risk of arrhythmia, and the FDA recommends elderly patients take only 20 mg a day, whereas 30 mg was found to reduce agitation in AD. Other researchers claimed that in general, too many meds are given to end-stage AD patients (see Sep 2014 news story). Last year brought bad news for PBT2, a compound Prana Biotechnology is developing, when its latest Phase 2 trial was negative (see Apr 2014 news story).
Given how long Phase 3 AD trials take to read out, several continued through 2014 without new announcements. For example, FORUM Pharmaceuticals (formerly EnVivo) continued testing an α7 nicotinic acetylcholine receptor agonist, Encenicline, and Lundbeck a serotonin receptor agonist called Idalopirdine. The TOMMORROW study is testing whether variants in the Tomm40 gene predispose to AD and if the insulin sensitizer pioglitazone can prevent it.
Interest in drugs targeted at tau grew considerably in 2014. They included Phase 3 trials of LMT-X, a methylene blue derivative purported to prevent tau aggregation in people with AD or frontotemporal dementia, as well as a new therapeutic vaccine, ACI-35, in Phase 1. More broadly, 2014 saw FTD research blossom with presentations at the International Conference on Frontotemporal Dementias revealing how far the field has come toward understanding the pathology, genetics, clinical presentation, and other aspects of the disease (see Nov 2014 conference series). FORUM Pharmaceuticals' HDAC inhibitor FRM-0334 moved into Phase 2 testing in patients with FTD caused by progranulin mutations (see Nov 2014 conference story).
There was a renewed call for combination therapies. A trial of vitamin E as an adjunct to memantine returned a small functional benefit, and a combination of two approved medications is underway in France (see Jan 2014 news story; Dec 2014 conference story).
Perhaps the most radical argument in 2014 about how to speed up the search for better medications came from Rusty Katz, formerly of the FDA. Katz urged the field to focus squarely on achieving large therapeutic effects, even to the point where trials build in futility analyses that set a high bar so that drugs which achieve a small therapeutic benefit get discontinued early in favor of putting efforts toward other drugs or combinations of investigational drugs that might make a much bigger impact. This, Katz said, is the way to realize the treatment goals of the U.S. National Plan to Address Alzheimer’s Disease. Thus far, the AD field has mostly paid lip service to the idea of combination trials (see Mar 2014 conference story). Katz reminded trialists that regulatory hurdles are not the holdup, as FDA guidance is in place (see Dec 2014 conference story).
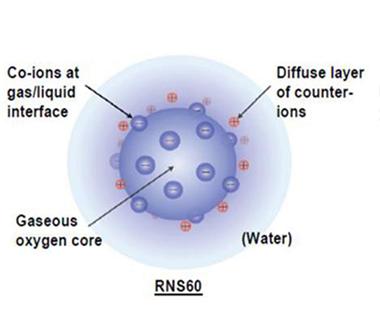
Powerful Packages.
Charged ions line the surface of nanobubbles filled with oxygen in this depiction of the structure of RNS60. [Image courtesy of Roy et al., PLOS One, 2014.]
In the year's curiosity department, researchers at AFFiRiS AG claimed that their placebo worked better than their active vaccine (see Jun 2014 news story), while a small company in Tacoma, Washington, said it planned to test nanobubbles, teeny packets of oxygen, in AD trials soon (see image at right; Dec 2014 conference story).
Others posit that exercise, specific diets, and maintaining cardiovascular health may work as well as drugs. Resveratrol, cocoa flavanols, and control of midlife high blood pressure were all reported to protect against cognitive decline (see Jun 2014 news story; Oct 2014 news story; Aug 2014 news story). The FINGER trial underway in Finland offered the strongest evidence to date that lifestyle intervention can improve cognitive function, and perhaps protect against AD (see Jul 2014 conference story).
With the field pushing to test interventions earlier in the disease progression, finding suitable tests to identify patients and measure their progress has been a prime challenge, but 2014 brought much progress there. In June, researchers reported that the ADCS Preclinical Alzheimer Cognitive Construct might fit the bill. Over three years, scores on the ADCS-PACC dropped in "cognitively normal" people who had amyloid in their brain (see Jun 2014 news story). The ADCS-PACC is being used in the A4 secondary prevention trial, which dosed its first patient in June 2014. Other public-private secondary prevention trials that are up and running, such as DIAN and API, have developed their own, similar composites tapping into the domains of episodic memory, attention, and executive function (see Dec 2014 conference story).
In its effort to open the door to a CSF-based diagnostic for early AD, the Global Biomarkers Standardization Consortium made a step when it published a standardized reference protocol for measuring cerebrospinal fluid Aβ42 (see May 2014 news story). The goal is a certified test that performs robustly in multicenter trials and even routine clinical practice worldwide.
Brain Imaging
In 2014, the field sprinted toward an approved diagnostic tracer for in vivo imaging of tau fibrils in the brain. Eli Lilly's T807 and two classes of compound being developed in Japan, THK-5117 from Tohoku University and PBB3 from the National Institute of Radiological Sciences, Chiba, all look like suitable PET ligands for human tau (see May 2014 conference story; Aug 2014 conference story). Binding of T807 in the brain jibes with the Braak staging pathologists have described in postmortem study, as well as with the areas responsible for cognitive deficits in AD. It also appears to show that tau tangles are limited to the medial temporal lobe during aging, but spread to the cortex when amyloid pathology kicks in (see Dec 2014 conference story).
In Aβ imaging, florbetaben joined florbetapir and flutemetamol in the ranks of approved imaging ligands. Regulatory agencies have defined their use such that they serve to exclude an AD diagnosis in people who test negative for the tracer, however, diagnosing physicians are only slowly beginning to subjugate their clinical impressions to amyloid imaging (see May 2014 news story; Dec 2014 conference story). Amyloid imaging in 2014 was in a transition phase, with researchers gathering data to show its usefulness in clinical practice in the hopes that the Centers for Medicare & Medicaid Services will reconsider and recommend reimbursement of amyloid PET scans more broadly in clinical settings.
Meanwhile, data from large prospective studies confirmed that people with amyloid in their brains are much likelier to decline cognitively, and research is now focusing on understanding the factors, such as ApoE genotype or cardiovascular comorbidities, that determine how quickly this happens (see Dec 2014 conference story).
Last but by no means least, researchers may have finally cracked the door open on imaging neuroinflammation. GE180, a fluorine-18 ligand that binds a mitochondrial transporter protein and labels activated microglia in mouse brain, is being tested in healthy volunteers and in people with AD and relapsing/remitting multiple sclerosis (see Dec 2014 conference story). Look to data from those trials in 2015.
Biomarkers
Researchers continue to seek alternative biomarkers to expensive PET imaging and invasive cerebrospinal fluid analysis. At the Alzheimer's Association International Conference in July, researchers reported that they can detect Aβ in the retina using a fluorescent dye and a specialized camera. Some scientists believe this may be a more reliable diagnostic than an eye lens-based test, since blood vessels innervate the retina where they may deliver and clear Aβ much like they do in other parts of the brain (see Jul 2014 conference story).
A blood-based test for AD seems no closer to reality despite much effort in this area. A new twist on that approach emerged in the form of lipid analysis. While many labs have analyzed the blood proteome to identify an AD signature, researchers reported in March that plasma phospholipids may hold the key. A panel of 10 blood lipids predicted which patients would develop cognitive problems during the next two to three years (see Mar 2014 news story). It remains to be seen if this panel specifically detects early AD or other neurodegenerative diseases. Many previous panels of factors in the blood have not translated into diagnostics.
Tau
Researchers surprised the AD field in March when they reported that in the DIAN longitudinal study of families with early onset AD, tau in the cerebrospinal fluid tracked lower over time once people showed symptoms of cognitive impairment (see Mar 2014 news story). Earlier cross-sectional studies had predicted that tau would continue to creep up in the CSF as the disease progressed, but this study, along with a few others, made scientists rethink how to use tau as a biomarker in efficacy trials. CSF tau is no longer considered a dynamic marker of progression. Tau has long been considered a marker of neurodegeneration, released by dead or dying cells, but its story became much more complex, and interesting, when researchers discovered that normal neurons appear to release the protein when stimulated (see Feb 2014 news story). Others reported that tau, a microtubule protein found in axons and soma, might play a physiological role in dendrites. In response to electrical stimulation, tau appeared to promote synaptic plasticity in dendrites by modifying the actin cytoskeleton (see Apr 2014 news story).
Scientists are trying to identify the most toxic forms of tau. One group reported a cell-based assay that detects species that promote aggregation of the protein (see Oct 2014 news story). This biosensor detected tau seeds in transgenic mice long before tau aggregated in the animals' brains. Extracts from human AD brain tissue, but not CSF, also tested positive in this test. The researchers do not yet know if the tau seeds they can measure are toxic.

Seed Sensors.
Tau inclusions (green, right) speckle tau biosensor cells one day after treatment with tau fibrils. FRET signals in these cells detect seeds at femtomolar concentrations. [Image courtesy of Holmes et al., PNAS 2014.]
Tau undergoes complex processing steps in neurons, being phosphorylated, acetylated, and cleaved by a variety of proteases. Add asparagine endopeptidase to the latter mix. Researchers reported that knocking out this peptidase prevents cleavage of tau in several places and protects transgenic mice that express mutant forms of human tau (see image at left; Oct 2014 news story). Similarly, others reported that blocking calpain proteases protects mice from tau toxicity (see Jul 2014 news story), and a small-molecule calpain inhibitor has finally entered Phase 1 testing for AD. Pharmaceutical companies are intrigued by tau glycosylation and are testing inhibitors of the glycoside hydrolase O-GlcNAcase in preclinical experiments. By blocking removal of sugars from tau, these small molecules may prevent tau from forming oligomers or larger aggregates. The year also saw progress in the development of active and passive tau immunotherapies (see Jul 2014 conference story).
Aβ
In 2014, the momentum of mechanistic research in neurodegenerative diseases shifted away from Aβ in favor of tau and other misfolding proteins. No AD research roundup would be complete without a mention of Aβ, however. In February, the link between the endocytic receptor SORLA and AD pathology grew stronger when researchers found genetic variants in SORLA that associate with early onset AD slowed Aβ clearance (see Feb 2014 news story). The G511R mutation of SORLA failed to escort Aβ to the lysosome for degradation. In related news, small molecules that stabilize the retromer appeared to quell Aβ production in neurons. The retromer is a protein machine that whisks APP out of endosomes and away from the influence of β-secretase. In neurons, the retromer stabilizer R55 steered APP toward non-amyloidogenic processing via α-secretase (see Apr 2014 news story).
In 2015, make sure to get your beauty sleep. Evidence continued to build last year that the brain drains Aβ through the glymphatic system, and that this happens best when we sleep and less well as we age. Other support grew for the idea that sleep disturbances put people at risk of AD, as well (see Oct 2014 news story; May 2014 conference story).
The field still struggles to identify the most toxic form of Aβ in the brain. A carbon fiber microelectrode that can detect Aβ in the brain debuted at the Society for Neuroscience meeting in Washington, D.C. (see Nov 2014 conference story). The device can measure concentrations of the peptide every 30 seconds, giving researchers a tool to measure rapid fluctuations in Aβ levels. It potentially could be modified to measure specific Aβ species or other proteins, such as tau.
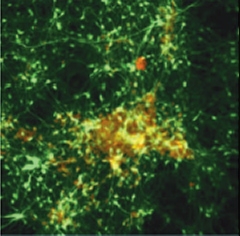
Deposition in a Dish.
After six weeks in three-dimensional gel culture, neurons (green) expressing familial Alzheimer's mutations form extracellular amyloid deposits (red/yellow). [Image courtesy of Choi et al., Nature 2014]
A cell culture model introduced in 2014 might help researchers pinpoint toxic Aβ species. This model recapitulates the two hallmarks of AD, amyloid plaques and neurofibrillary tangles, in a three-dimensional, gel-based neuron culture (see image at right; Oct 2014 news story). One downside is the model relies on overexpression of APP, a criticism of most of the animal models used for preclinical studies. Two new knock-in APP models that develop amyloid plaques, gliosis, synaptic loss, and cognitive impairment without overexpressing the precursor protein may help with translational research (see Apr 2014 Webinar). A BACE1 knock-in model debuted as well. It also develops some of the hallmarks of AD without overexpressing APP or BACE1 (see Aug 2014 news story).
Genetics
Although geneticists are largely done conducting genome-wide association studies in AD, they continue to glean information from the data, reporting that some GWAS hits not only predispose to AD but accelerate its onset. Others linked GWAS hits to epigenetic changes that may regulate the expression of nearby genes (see Nov 2014 news story; Nov 2014 news story). Scientists have developed different methods to find new genes that increase risk for AD. While GWAS identify commonly inherited sequences, they miss rare variants. For example, a family based approach using whole-genome sequencing identified a single nucleotide polymorphism in the UNC5C gene that predisposes to late-onset AD and potentially other neurodegenerative diseases. UNC5C encodes a protein that promotes apoptosis, or programmed cell death, and the single amino acid change seems to keep the protein turned on. Others have adopted whole-exome sequencing to find variants that might protect against AD in the people most susceptible to it, namely those who are homozygous for ApoE4 (see Dec 2014 conference story). In a widely praised study, researchers combined exome sequencing with network analysis to find rare genetic variants that cause hereditary spastic paraplegia. They linked those variants to common biological pathways that underlie neurodegenerative diseases, including AD (see Jan 2014 news story). Pundits predicted that similar approaches could be used to identify AD-specific genes.
C9ORF72
Hexanucleotide repeat extensions in intron 1 of C9ORF72 underlie a quarter of familial FTD cases and about 40 percent of familial ALS cases. In 2013, researchers reported that the intron is transcribed and translated into dipeptide repeats in both sense and antisense directions. Scientists were left to puzzle over which of five potential poly-dipeptides and two RNAs with expanded repeats might explain why this mutation is toxic. Last year brought ample data, but no closure on this question. Some researchers implicated arginine-rich dipeptide repeats (see Aug 2014 news story and Dec 2014 news story), but others favored the RNA (see Nov 2014 conference story). The guanine-rich hexanucleotides in mutant C9ORF72 form structures called G-quadruplexes. These may interfere with similar quadruplexes formed when angiogenin cleaves transfer RNAs, which appear to promote RNA/protein granules that protect cells from stress (see Dec 2014 news story). Expansions in C9ORF72 cropped up in multiple-system atrophy and pseudodementia, though their involvement in the pathology of these disorders remains uncertain (see Apr 2014 news story). Researchers investigated small molecules that bind RNA hexanucleotide repeats as a potential treatment for ALS/FTD, and one dipeptide repeat as a potential biomarker in cerebrospinal fluid (see Aug 2014 news story).
TREM2 and Microglia
After ApoE4, variants in TREM2 are the strongest genetic risk factors for late-onset AD. While the biology behind this association remains murky, 2014 saw both advances and surprises in TREM2 research. In June, researchers revealed that knocking out one copy of this cell surface receptor limited the number of microglia surrounding amyloid plaques in the cortex of APP/PS1 models of AD. Curiously, this dearth of microglia had no effect on plaque burden (see Jun 2014 news story). Others repeated this data, reporting that mice lacking both copies of the TREM2 gene mounted little microglial response around plaques in APP/PS1 mice and that plaque numbers in the cortex were similar to those in TREM2-positive control animals (see Nov 2014 conference story). Experts in the field are unsure what to make of these data, and rumor has it that other labs have seen greater plaque numbers in TREM2 knockouts. Add to this confusion over which cells make TREM2. At the Society for Neuroscience meeting, researchers claimed that cells in the brain that express this receptor are not brain-resident microglia, but monocytes that infiltrate from the periphery.
Researchers finally got a handle on the function of TREM2, reporting that mutations linked to neurodegenerative diseases scupper phagocytosis. The problem seems rooted in a breakdown in TREM2 processing at the cell surface. The extracellular domain of the protein is shed by α-secretases, much like the soluble extracellular domain of APP. The intramembrane stub goes on to be processed a la Notch and APP C-terminal fragments. The mutations trap TREM2 in the endoplasmic reticulum, the protein never matures, and the extracellular domain never sheds (see Jul 2014 Webinar on Kleinberger et al., 2014). The shed part of TREM2 ends up in the cerebrospinal fluid, but not in people who carry the disease mutations. Interestingly, some people with AD who have normal copies of TREM2 have less of the soluble domain in the CSF, hinting that the fragment might be a valuable diagnostic marker for weak microglial function. In early 2014 geneticists reported that the R47H TREM2 variant associated with AD also doubled the odds of developing amyotrophic lateral sclerosis (see Feb 2014 news story).
In 2014 researchers reported that microglial progenitors exist in the brain. This came as a surprise since microglia are believed to arise during development from the yolk sac. Theory had it that cells from this sac made their way to the brain where they became microglia, and then were cut off from other immune cells as the blood-brain barrier formed. But researchers reported that after wiping out all microglia in the brain, their numbers bounced back within two weeks, and the new cells seemed to originate from within the brain (see Apr 2014 news story). The progenitor cells tested positive for nestin, a neuronal marker, suggesting a wholly unrecognized source of microglia in the brain. In other microglial news, researchers found that when these cells lack progranulin, they fail to clear up deposits of Aβ (see Oct 2014 news story). This could explain why mutations in the progranulin gene increase the risk for Alzheimer's.
Connectivity Networks and Neurodegeneration
The last decade has seen evidence grow that Alzheimer's is as much a disease of cortical neural networks as of neurodegeneration. For example, while the hippocampus atrophies in both semantic dementia and AD, only people with the latter have episodic memory problems. Researchers traced those memory issues to a breakdown in connectivity between the hippocampus and the posterior cortex, rather than to local hippocampal atrophy (see Mar 2014 news story). Similar breakdowns in neural connectivity in both familial AD and late-onset disease strengthened the idea that Aβ pathology underlies both (see Aug 2014 news story). In keeping with this, others found that Aβ causes hyperactivation of the entorhinal cortex during memory tasks and that the frontoparietal cortex begins to atrophy early in AD, just as Aβ in the CSF begins to fall, a sign the peptide has begun to accumulate in the brain parenchyma (see May 2014 news story; Aug 2014 news story). Some investigators have devised therapies to correct network failures. Transcranial magnetic stimulation designed to improve connections between the hippocampus and other brain regions reportedly strengthened associated memories in healthy volunteers, hinting that one day it may help AD patients remember (see Aug 2014 news story).

The Fabric of a Synapse. A section of a synaptic bouton, containing 60 different proteins, shows crisscrossing actin filaments (purple, from top), and vesicles (cream, right) that travel toward the synapse (bottom, orange). [Image courtesy of Wilhelm et al., 2014, Science.]
Scientists studying human brain connectivity hope to learn much from the Brain Activity Map Project launched in 2013. In the meantime, they are learning more about how the mouse brain is wired, and this should help them better characterize rodent models of human diseases. In the spring, scientists were treated to a high-resolution map of the mouse brain connectome and to the most detailed look yet at the synapse, reporting that a single one contains 300,000 proteins of about 1,000 different types (see image above) (Apr 2014 news story; May 2014 news story). A whole-body clearing method allowed researchers to visualize entire networks of cells in the mouse, including neurons in the brain and spinal cord (see Jul 2014 news story). Last but not least, a reconstruction of a famous brain in neuroscience promised to shed new light on human memory. Henry Molaison, known to researchers as H.M., died in 2008 and last January scientists published a three-dimensional digital reconstruction of his brain (see Jan 2014 news story). In 1953 Molaison had had much of his hippocampus removed to prevent uncontrollable seizures. He was 27. Post-surgery anterograde memory problems presented scientists with their first solid evidence that the hippocampus was essential for memory, and this led to a wealth of research. The three-dimensional reconstruction revealed that the surgery had left much more of the hippocampus intact than had been believed.
Funding and Global Coordination
While researchers continued to lament a poor NIH payline and insufficient funds to support the National Plan, there was a modicum of good news. Biomarkers Across Neurodegenerative Diseases, or BAND, announced $2 million in grants to mine data from the Alzheimer's Disease Neuroimaging Initiative and the Parkinson's Progression Markers Initiative (see Feb 2014 news story). The Accelerating Medicines Partnership, a collaboration among the NIH and 10 industry partners, promised $129.5 million to study biomarkers and therapeutic targets in AD (see Feb 2014 news story). Congress passed a budget, signed by President Obama, that slated an additional $80 million for the NIA, most of which was to go to fund the National Plan. Perhaps the coolest funding news was the ice bucket challenge, a simple idea that went viral and raised more than $100 million for ALS research (see Sep 2014 news story). In Europe, the Innovative Medicines Initiative slated €64 million for the European Prevention of Alzheimer's Dementia Consortium (see Dec 2014 conference story). EPAD will help select at-risk people for secondary prevention trials and assess which interventions should move forward for further testing. Globally, the World Dementia Council, which grew from U.K. Prime Minister David Cameron's presidency of the G8 in 2013, continued its efforts to coordinate initiatives to accelerate and improve therapy development.—Tom Fagan and Gabrielle Strobel
References
News Citations
- Crenezumab Disappoints in Phase 2, Researchers Remain Hopeful
- Immunotherapy I: Baby Steps, but No Breakthroughs
- End of the RoAD for Gantenerumab? Roche Declares Prodromal Alzheimer’s Trial Futile
- Denied Breakthroughs, Researchers Keep Hunting New Therapies
- Novartis to Partner with Banner Health on ApoE4 Prevention Trial
- Brain Shuttle Ferries Antibodies Across the Blood-Brain Barrier
- Less Is More: High-Affinity Antibodies Block Blood-Brain Barrier Conduit
- Antibody Ferry Looks Safe in Monkeys, Charts Course for Human Studies
- Lilly Teams Up With AstraZeneca for BACE Inhibitor Phase 2/3 Trial
- Research Brief: New BACE Inhibitor Joins the Fold
- Scant α-Secretase Compensation for BACE Inhibition in Monkeys
- At High Doses, BACE1 Inhibitors Hinder Synaptic Plasticity in Mice
- Citalopram Calms Agitation in Alzheimer’s, but Carries Risks
- A New Drug to Calm Agitation, Uncontrollable Laughing and Crying, in Alzheimer’s?
- Are Too Many Meds Given to End-Stage Dementia Patients?
- PBT2 Takes a Dive in Phase 2 Alzheimer’s Trial
- Progranulin-Boosting Drug Moves into Phase 2 for Frontotemporal Dementia
- Trial Suggests Vitamin E Protects Function in Mild Alzheimer's
- New Ideas for Alzheimer’s Treatment: What’s on Offer in 2015?
- Combination Trial Debate Energizes Keystone Symposium
- Rusty Unleashed: Forget Disease Modification, Go for Big Effect
- In Surprise, Placebo, not Aβ Vaccine, Said to Slow Alzheimer’s
- Nanobubbles: Potent Potion, or Just Effervescence?
- Resveratrol Improves Memory in Overweight Adults
- Cocoa Flavanols Give Memory a Boost
- Treating Midlife Hypertension Helps Preserve Cognition in Old Age
- Healthy Lives, Healthy Minds: Is it Really True?
- Test Battery Picks Up Cognitive Decline in Normal Populations
- From Shared CAP, Secondary Prevention Trials Are Off and Running
- More Accurate Ways to Measure CSF Aβ Debut
- PET Tracers Enlighten at Neurology Conference
- Scan by Scan, Growing Tau PET Data Picks Up Early Memory Deficits
- New Target Has Legs: Tau PET, Mice, and Antibodies
- Three’s Company: Florbetaben Approved, Excludes AD Diagnosis
- CTAD Shows Alzheimer’s Field Trying to Reinvent Itself
- Large Studies Agree: Brain Amyloid Accelerates Cognitive Decline
- Meet GE180: A PET Ligand for Tracking Neuroinflammation?
- Alzheimer’s Disease: In the Eye of the Patient?
- Do Lipids Hold the Key to Blood-Based Alzheimer’s Test?
- DIAN Longitudinal Data Surprises With Late Drop in Tau
- Neurons Release Tau in Response to Excitation
- Does Dendritic Tau Promote Plasticity?
- Cellular Biosensor Detects Tau Seeds Long Before They Sprout Pathology
- Does Novel Tau Protease Promote Pathology?
- Reining in Calpain Tempers Tau and Delays Neurodegeneration
- Therapies Take Aim at Tau
- SORLA Serves Up Aβ for Destruction
- Could Bolstering the Retromer Thwart Alzheimer’s?
- Wake Up and Smell the … Orexin? Peptide Percolates in Alzheimer’s Brain
- Glymphatic Flow, Sleep, microRNA Are Frontiers in Alzheimer’s Research
- Measuring Rapid Changes in Brain Aβ in Live Mice
- Alzheimer’s in a Dish? Aβ Stokes Tau Pathology in Third Dimension
- New BACE1 Mouse Models AD Without APP Overload
- Epigenetic Alterations Mark Alzheimer’s Disease Genes
- Do Alzheimer’s GWAS Hits Speed Up Disease?
- Scientists Seek, Find, and Exploit AD Risk Genes
- Exome–Network Combination Uncovers New Disease Genes
- C9ORF72’s Dirty Work Done by Problem Proteins
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
- Guanine-Based RNA Tetrads Turn on Stress Granules, Rescue Neurons
- C9ORF72 Repeats Expand into New Disorders—Cause, or Coincidence?
- Drug and Biomarker Candidates for C9ORF72 ALS and FTD
- TREM2 Mystery: Altered Microglia, No Effect on Plaques
- TREM2 Data Surprise at SfN Annual Meeting
- TREM2 Variant Doubles the Risk of ALS
- Microglial Magic: Drug Wipes Them Out, New Set Appears
- Does Progranulin in Microglia Protect Against Alzheimer’s?
- Cognitive Deficits Arise From Network, Not Regional, Dysfunction
- Neural Circuitry Goes Haywire in Both Sporadic and Familial AD
- Do Frontoparietal Regions Shrink First in Preclinical Alzheimer’s?
- Do Earliest Cognitive Deficits in Alzheimer's Appear in the Entorhinal Cortex?
- Boosting Memory Through Stimulation of a Brain Network
- A High-Resolution Wiring Diagram of the Mammalian Brain
- Synaptic Metropolis—Microscopic Census Documents 300,000 Protein Inhabitants
- Transparent Bodies Allow Neural Networks to ‘Apparate’
- A 3-D View of One of Neuroscience's Most Famous Brains
- Grants to Explore Boundaries Between Alzheimer’s and Parkinson’s
- New Initiative AMPs Up Alzheimer’s Research
- After a Summer of Icy Showers, What Will Happen with Buckets of Cash for ALS?
Conference Coverage Series Citations
Webinar Citations
- Good-Bye Overexpression, Hello APP Knock-in. A Better Model?
- Mutations Impair TREM2 Maturation, Processing, and Microglial Phagocytosis
Paper Citations
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Other Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.