Tau Roundup: Inducible Mice Accentuate Aggregation and More
Quick Links
It was 2 years ago that the ARF news desk noted a resurgence of interest in tau (see ARF related news story). Of course, tau was never really gone, but for a period, its role in AD pathology was overshadowed by an intense research focus on amyloid-β. The evidence of tau’s return was the mushrooming number of presentations on tau and Alzheimer disease at the 2005 Society for Neuroscience meeting. In the following years, interest in tau has continued to mount, as the protein, a neurodegenerative agent in its own right, has been implicated as a downstream effector of Aβ pathology (see ARF related news story) and as an increasingly compelling target for AD therapies (see ARF related news story).
A batch of new studies continues the trend. First, two new transgenic tau mouse models from the labs of Eva-Maria and Eckhard Mandelkow at the Max Planck Unit for Structural Molecular Biology, Hamburg, Germany, point a finger squarely at the ability of tau proteins to form a β-structure and aggregate as a primary determinant of their toxicity. The same lab has some new aggregation inhibitors in case any companies are willing to bite on the promise of anti-tau therapies. In other work, Erika Holzbaur and colleagues at the University of Pennsylvania in Philadelphia give a close-up look at how excess tau inhibits microtubule-dependent transport, a deadly roadblock that can lead to synapse loss.
In Alzheimer disease brain, the tau protein is subject to a number of insults: it is evicted from its normal home in the axon and becomes spread throughout the cell, and it is subject to post-translational modifications including phosphorylation at more than a dozen sites as well as proteolytic cleavage. Eventually, tau aggregates and produces neurofibrillary tangles, a hallmark of AD pathology. Just where in this mess it expresses toxicity is not clear.
To get at the role of aggregation, Maria-Magdalena Mocanu and Astrid Nissen from Eva-Maria Mandelkow’s lab produced transgenic mice with inducible and brain-restricted expression of a human tau fragment (residues 244-372), spanning the repeat domain region responsible for microtubule association. They compared two mutants, one the highly aggregating δK280, the other a non-aggregating version of the same peptide, in which two proline substitutions prevent formation of the β-structure necessary for aggregation. Their paper, published in January 16 Journal of Neuroscience, shows that the mice with the “proaggregation” peptide develop tau aggregates and tangles within 3 months after the gene is turned on. Astrogliosis, synaptic loss, and neurodegeneration appear from 3-5 months onward. This occurs even with low expression of the protein, which accumulates to only about 70 percent of the level of endogenous mouse tau. In contrast, the “antiaggregation” peptide, expressed at equivalent levels, shows no such effects even after 22 months. The results suggest that neurodegeneration stems from neurotoxicity due to aggregation, not merely the expression of human tau.
The new paper follows a report in the JBC last October, where the same group, with first author Katrin Eckermann, produced similar results in mice expressing full-length tau with the same mutations. They found that while both mutants become mislocalized and hyperphosphorylated to some extent, only the proaggregation mutant appears in the insoluble fraction of brain extracts and adopts a pathological conformation as detected by the MC1 antibody. As expected from in vitro studies, the full-length proteins take much longer than the repeat fragment to form tangles, but some tangles are seen in very old mice (22 months). Nonetheless, synaptic loss was detected in mice by 13 months of age. These results further support the idea that the propensity of the protein to form β-sheets determines neurodegeneration, as the full-length protein has a much lower β-tendency than the repeat domain fragment. This parallels some recent studies on Aβ mutants, which showed a similar correlation between the tendency of different protein sequences to form β-sheet structures and neurotoxicity in fruit fly models (see ARF related news story).
With the generation of a number of mouse strains that express human tau, one question in the field has been whether endogenous mouse tau participates in the aggregates induced by expression of various forms of human tau. Because the human tau species expressed in the new mice are easily distinguished from mouse tau by their size, the investigators were able to produce clear evidence of the mislocalization and eventual incorporation of mouse tau into neurofibrillary tangles under the influence of the human protein. This observation “settles a longstanding debate in the field over whether or not coaggregation occurs in vivo,” the Mandelkows told Alzforum in a recent conversation that ranged widely over the subject of tau and AD. It also has ramifications for human disease, since it suggests that once a nucleation event occurs around abnormal protein, normal tau can be recruited into a pathological conformation as well.
This mechanism might account for the spreading of neurofibrillary tangles in cells, Eckhard Mandelkow noted. “If you have a species of tau that for some reason has a higher tendency to aggregate, either because it’s complexed with some other factor, or because it’s cleaved to a fragment that has a higher tendency to aggregate, then this center of nucleation can spread.” On the other hand, he said, the results indicate that the process can be inhibited or reversed.
The recruitment of normal tau may also explain the observation that neurofibrillary tangles persist when the human tau gene is turned off in the new mice. In mice expressing truncated tau, switching off the transgene resulted in a clearance of the human protein within 6 weeks. Both soluble and aggregated human tau disappeared, but tangles, hyperphosphorylation, and missorting of the mouse protein persisted. Thus, it appears that mutated and aggregated tau is readily degraded, while normal mouse tau is continuously recruited to take its place in the tangles. The finding of persistent tangles jibes with another inducible tau mouse model from Karen Ashe and colleagues. They found tau expression, neuronal loss and memory effects were all reversible by turning off expression of a mutated tau transgene, but the tangles remained and continued to accumulate (see ARF related news story).
The Mandelkows also looked at reversibility of tau toxicity in their mice. Turning off tau transgene reduced astrogliosis, but the mice still displayed changes in neuron morphology and neuron loss in regions that had neurofibrillary tangles. Eva-Maria Mandelkow told ARF that they have their first data showing behavioral deficits and effects on long-term potentiation in the mice, and the reversal experiments are now underway. They will know much more on this count in the next 6 months, she said.
As for the identity of the toxic species, that is an open question. Eva-Maria Mandelkow says. “We are working on oligomers—they are in at the moment—but it’s not clear yet that it’s oligomers.” What is clear, she says, is that the neurotoxicity of tau comes from its propensity to form a β-structure and aggregate. “Now we know that once you get rid of aggregates, you can get rid of toxicity. We see this wherever we test it, whether in the test tube or in neuronal cell models or now in mice. With the proline mutations or when we treat with antiaggregation compounds, the cells are fine. So that is what we are thinking of when we think of therapies.”
Tau and Transport
One of earliest signs of AD on the neuron level is the redistribution of tau from axons into the cell body and dendrites, which may cause another toxic action of the protein, the inhibition of microtubule-mediated axonal transport. A paper in the January 17 Science from the Holzbaur lab shows at the single molecule level, encounters between tau deposits on microtubule tracks and single dynein or kinesin motor complexes in vitro inhibits the progress of both motors. From previous work of the Mandelkow lab, they knew that tau on microtubules inhibited kinesin both in vivo and in vitro (Seitz et al., 2002), and this accounts for the inhibition of anterograde axonal transport by the protein.
In the new work, first author Ram Dixit shows that tau directly inhibits dynein motor movement as well. But the inhibition is not the same as for kinesin: when kinesin encounters tau on microtubules, it tends to fall off, whereas dynein, they show, tends to reverse direction. In addition, they show that kinesin is much more sensitive to tau interference than dynein.
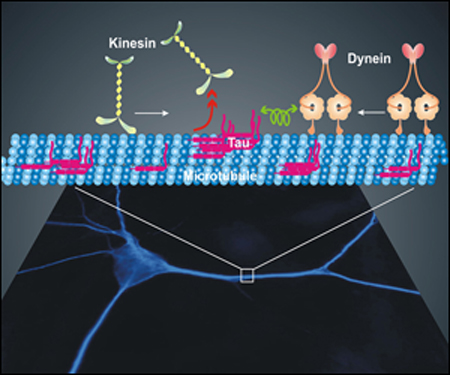
Tau Trouble on the Tracks
Imaging of single kinesin or dynein motors as they encounter tau on microtubule tracks shows that both are inhibited, but in different ways. Kinesin reacts to tau by falling off the track, while dynein reverses direction. Image credit: the Holzbaur lab.
The work shows how the elevation of tau in the cell body and dendrites seen early in AD might inhibit transport, simply by taking up more of the microtubule and blocking the motor’s path. The researchers describe how the normal gradient of tau along the axon might promote the loading of kinesin and cargos at the cell body end (low tau) and then the unloading at the distal, synaptic end (high tau). Dynein-mediated retrograde transport would not be affected at physiological tau concentrations. “Based on this model,” they write, “perturbing tau distribution would predictably impair axonal transport. For example, in Alzheimer’s disease, tau accumulates in the somatodendritic compartment. Our model then predicts that kinesin-driven anterograde transport of new material to the axon terminal would be severely compromised, leading to neurodegeneration.”
Indeed, this is precisely what was shown last year in in vitro studies by Edda Thies and Eva-Maria Mandelkow (Thies and Mandelkow, 2007). In that work, when they overexpressed tau in cultured hippocampal neurons, they saw the misdirection of the protein into the somatodendritic compartment and degeneration of synapses. The mechanism was transport inhibition of vesicles and organelles, leading to mitochondrial degeneration. The tau-induced synaptic decay they observed was prevented by activation of the kinase MARK2 (microtubule-associated protein/microtubule affinity regulating kinase 2), which phosphorylated tau, removed excess tau from the microtubule tracks and reversed the transport block.
The relationship between inhibition of transport, early synapse loss, post-translational modifications like phosphorylation or cleavage, the aggregation of tau, and ultimately, neurodegeneration, are not at all clear. But early (and reversible) synapse loss occurs in response to Aβ, and the results raise the possibility that tau may have a role in that loss, even before plaques or tangles appear.
Finally, another recent paper from the Mandelkows provides an interesting explanation for how tau, which is normally associated with microtubules, can spread itself all over the cell even as it blocks transport. A report from Sven Konzack, published in the Journal of Neuroscience last fall, shows that tau, in fact, can diffuse very quickly through cells. Using live cell fluorescence microscopy, Konzack found that tau was not just stuck on microtubules, but moved around quite freely. This suggested that tau was rapidly moving on and off microtubules. If phosphorylated, its movements became independent of microtubules altogether.
“This rapid diffusion is important because of the early redistribution of tau to the somatodendritic compartment in AD," Eckhard Mandelkow explained. “From our perspective, the question is not why tau is in axons only, but why it is not everywhere all the time because it can move so easily. Some people believe there is a special axon targeting mechanism for tau, or there could be selective degradation in other compartments. One of these mechanisms breaks down in AD, and you get improper distribution.”
“That could have something to do with phosphorylation," Eva-Maria Mandelkow added.
Treating Tau
Two prominent avenues of investigation to find tau-directed therapies involve inhibitors of aggregation and modulators of phosphorylation (with the assumption that phosphorylation is a prelude to aggregation). In speaking with Alzforum, the Mandelkows highlighted their early and continuing interest in aggregation inhibitors as potential treatments for AD and tauopathies, and the group has actively pursued small molecules that block tau aggregation and toxicity in vitro and in cell-based assays (see ARF related news story and ARF news story). Five years ago they screened 200,000 small molecules, and their efforts have led to the discovery of novel inhibitors, as well as generating structural information that will aid with further inhibitor development (Pickhardt et al., 2007). Recent work in their lab by Bruno Bulic and Marcus Pickhardt, published in the Angewandte Chemie International Edition of November 5, presented a new class of potent rhodanine-based inhibitors (100-600 nM in cells) that not only prevent aggregation but also promote the disassembly of paired helical filaments. In addition to testing their best compounds on their own models, they are also hoping to find collaborators to move them forward into pharmacokinetic and animal testing.
There are other potential tau targets out there, too. In people with Alzheimer disease, tau is not mutated, and it is not clear what happens to render the protein toxic. One possible culprit could be cleavage by an unknown protease that results in a proaggregation peptide of tau that can seed fibril formation (see ARF related news story). That protease could be a target for therapy, and is another prospect the Mandelkow lab is pursuing. Not all agree on the cleavage angle, however. A recent paper from the lab of Michel Goedert concludes that a widely studied caspase cleavage of tau at D421 is unlikely to be necessary for assembly of tau into filaments (Delobel et al., 2008).
As long-time tau researchers, the Mandelkows join others in the field in their enthusiasm for the surge in interest in tau among Alzheimer researchers. “I am astonished how much interest there is all of a sudden related to tau issues in Alzheimer disease,” Eva-Maria Mandelkow told ARF. “I think it is good, because tau is the other component that is actually aggregating in the brain and killing the cells.”—Pat McCaffrey
References
News Citations
- SfN: Return of the Other—Tau Is Back, Part 1
- APP Mice: Losing Tau Solves Their Memory Problems
- Philadelphia: Targets in a Barely Tapped Market Keep Big Pharma Focused on AD
- Shaping Up Amyloid Toxicity: Does It Compute?
- No Toxicity in Tau’s Tangles?
- Paper Alert: Inducing and Reversing Cell-Based Tau Aggregation
- Does Tau Truncation Sow Seeds of Aggregation?
Paper Citations
- Seitz A, Kojima H, Oiwa K, Mandelkow EM, Song YH, Mandelkow E. Single-molecule investigation of the interference between kinesin, tau and MAP2c. EMBO J. 2002 Sep 16;21(18):4896-905. PubMed.
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007 Mar 14;27(11):2896-907. PubMed.
- Pickhardt M, Larbig G, Khlistunova I, Coksezen A, Meyer B, Mandelkow EM, Schmidt B, Mandelkow E. Phenylthiazolyl-hydrazide and its derivatives are potent inhibitors of tau aggregation and toxicity in vitro and in cells. Biochemistry. 2007 Sep 4;46(35):10016-23. PubMed.
- Delobel P, Lavenir I, Fraser G, Ingram E, Holzer M, Ghetti B, Spillantini MG, Crowther RA, Goedert M. Analysis of tau phosphorylation and truncation in a mouse model of human tauopathy. Am J Pathol. 2008 Jan;172(1):123-31. PubMed.
Further Reading
Papers
- Bolmont T, Clavaguera F, Meyer-Luehmann M, Herzig MC, Radde R, Staufenbiel M, Lewis J, Hutton M, Tolnay M, Jucker M. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP x Tau transgenic mice. Am J Pathol. 2007 Dec;171(6):2012-20. PubMed.
News
- News on Tau: More Mice Enter the Picture, Structure Takes Shape
- SfN: Return of the Other—Tau Is Back, Part 1
- APP Mice: Losing Tau Solves Their Memory Problems
- Philadelphia: Targets in a Barely Tapped Market Keep Big Pharma Focused on AD
- Paper Alert: Inducing and Reversing Cell-Based Tau Aggregation
- Does Tau Truncation Sow Seeds of Aggregation?
- Shaping Up Amyloid Toxicity: Does It Compute?
- No Toxicity in Tau’s Tangles?
Primary Papers
- Eckermann K, Mocanu MM, Khlistunova I, Biernat J, Nissen A, Hofmann A, Schönig K, Bujard H, Haemisch A, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The beta-propensity of Tau determines aggregation and synaptic loss in inducible mouse models of tauopathy. J Biol Chem. 2007 Oct 26;282(43):31755-65. Epub 2007 Aug 23 PubMed.
- Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008 Feb 22;319(5866):1086-9. PubMed.
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schönig K, Bujard H, Mandelkow E, Zhou L, Rune G, Mandelkow EM. The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy. J Neurosci. 2008 Jan 16;28(3):737-48. PubMed.
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007 Mar 14;27(11):2896-907. PubMed.
- Konzack S, Thies E, Marx A, Mandelkow EM, Mandelkow E. Swimming against the tide: mobility of the microtubule-associated protein tau in neurons. J Neurosci. 2007 Sep 12;27(37):9916-27. PubMed.
- Bulic B, Pickhardt M, Khlistunova I, Biernat J, Mandelkow EM, Mandelkow E, Waldmann H. Rhodanine-based tau aggregation inhibitors in cell models of tauopathy. Angew Chem Int Ed Engl. 2007;46(48):9215-9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.