α-Synuclein Spurs Neuroinflammation Via Microglial LRRK2
Quick Links
Scientists believe that in Parkinson’s disease and other synucleinopathies, toxic α-synuclein can escape the confines of damaged dopaminergic neurons, infiltrate neighboring microglia, and set off a neuroinflammatory storm. But what exactly happens inside the microglia? A paper in the October 14 Science Translational Medicine offers some clues. In mouse microglia, α-synuclein enhances the activity of another well-known agent of Parkinson’s disease, the leucine-rich repeat kinase 2, according to scientists led by Changyoun Kim and Eliezer Masliah from the National Institute on Aging, Bethesda, Maryland. LRRK2 then sets off a signaling cascade mediated by a transcription factor called nuclear factor of activated T cells, cytoplasmic 2. In turn, NFATc2 leads to a neuroinflammatory cascade and possibly many of the symptoms of Parkinson’s, dementia with Lewy bodies (DLB), and other synucleinopathies, according to the authors.
- α-Synuclein enters microglia through Toll-like receptor, activating LRRK2.
- LRRK2 then sets off a signaling cascade mediated by NFATc2.
- Microglia churn out inflammatory cytokines.
Zhenyu Yue, Icahn School of Medicine at Mount Sinai, sees this study as an important step forward in PD research. “This is really exciting work,” he told Alzforum.
Previously, scientists reported that α-synuclein, a normally cytoplasmic protein, can shimmy from neurons to other neurons and even to glial cells (Lim et al., 2018). Masliah and colleagues found that the synuclein can enter microglia via Toll-like receptor 2 on the cell surface and, once inside, it instills release of neuroinflammatory cytokines, including tumor necrosis factor-α (TNFα) and interleukin-6 (IL-6) (Kim et al., 2013; Kim et al., 2016). But what happens immediately after α-synuclein enters the microglia has been a mystery.
To investigate, Kim and colleagues dripped conditioned medium from neuronal cells expressing either human α-synuclein (aSCM), or β-galactosidase (LZCM) as a control, onto cultured mouse microglia. Zero, two, six, or 24 hours later, they analyzed whole microglia lysates for expression of inflammatory mediators, including LRRK2 and Nf-κB.
Previous research had linked this serine/threonine kinase to microglial inflammatory responses (Moehle et al., 2012; Marker et al., 2012). In the current study, exposure to aSCM, not LZCM, increased phosphorylation of LRRK2 itself at serines 935 and 955, nuclear factor κB (NF-κB) at serine 536, and p53 at serine 15 over time (see image below). Phosphorylation of LRRK2 substrates Rab-8A and Rab-10 also increased.

Synuclein Trips Microglia. Mouse microglia treated with αSCM turned up phosphorylation of LRRK2, NF-κB, and p53. [Courtesy of Kim et al., Science Translational Medicine 2020.]
To see how these responses might generate cytokines, Kim and colleagues analyzed the transcriptomes of rat microglia that had been exposed to αSCM. Among 213 upregulated genes, they found 43 that are likely to be involved in Toll-like receptor signaling, including NFATc2. A network analysis based on protein-protein interactions and gene function suggested that LRRK2 and NFATc2 directly interact. The authors found that LRRK2 both directly phosphorylates NFATc2 at threonines 483, 733, 862, 870, and 893, and mediates its translocation from the microglial cytoplasm to the nucleus, where it modulates gene expression. This shift into the nucleus activated NF-κB, which increased expression of TNFα and IL-6, leading to their release into the medium.
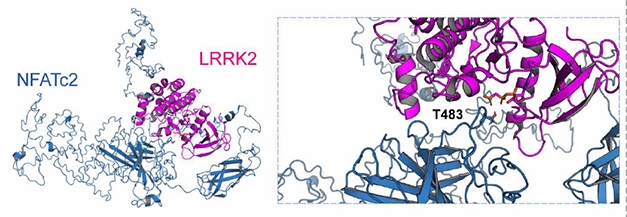
Partners in Inflammation? In silico modelling (left) based on the crystal structure of human NFATc2 (blue) and an amoeba homolog of LRRK2 kinase domain (purple) implies that LRRK2 can phosphorylate NFATc2 threonine 483 (insert). [Courtesy of Kim et al., 2020 Science Translational Medicine.]
Mouse cells, rat cells, amoeba LRRK2 domain—do people with a synucleinopathy have more nuclear NFATc2 than normal? To take their work back into humans, the scientists used postmortem data from the Alzheimer Disease Research Center at the University of California, San Diego, where people with synucleinopathies are also enrolled in research studies. They discovered more NFATc2 in the neocortex and striatum tissue of 10 people with either PD or DLB than in eight controls.
To test if NFATc2 ticks up in human microglia, the scientists plan to take cells from patients, derive induced pluripotent stem cells, and then use those to create microglia.
Seeing the neuroinflammatory pathway in mouse microglia more clearly, Kim and colleagues next inhibited LRRK2 in a mouse model of synucleinopathy to see if they could stop disease progression. Thy1-α-synuclein mice overexpress human α-synuclein and develop pathological features of Parkinson’s disease beginning when they are around 1 month old. Giving the LRRK2 inhibitor HG-10-102-01 to a dozen 9-month-old mice five days per week for four weeks improved the animals’ balance on a rotating rod, though nonmotor symptoms, such as the ability to hang from a wire cage, remained impaired. Moreover, the mice were even less anxious and more willing to explore open environments than untreated transgenic controls.
The authors believe that blocking the interaction between LRRK2 and NFATc2 may benefit people with PD, DLB, and multiple system atrophy (MSA). Alas, there’s a snag. Developing a therapy that targets LRRK2 in microglia, as opposed to in neurons, is currently not possible, said Masliah. “We must also worry about impacting LRRK2 function in other cells, including those in the kidney and lungs,” he told Alzforum.
In fact, LRRK2 inhibitors developed for clinical testing change the morphology of alveoli in the lungs of nonhuman primates, though this does not seem to compromise lung function (Apr 2020 news). Masliah suggests using a low dose, which, while hampering the protein in neurons, would have a stronger effect in microglia, where LRRK2 is more abundant. Drugs that target toxic α-synuclein will also be crucial, said Masliah. “It’s synergistic—the drugs need to be developed together.”
Yue noted, however, that NFATc2 is not the only LRRK2 kinase substrate. He wonders if others, such as Ras-related proteins Rab-8A and Rab-10, could also lead to downstream neuroinflammation. To fully understand the process, “This group, and others should look into the direct contribution of any other substrates of LRRK2 that might mediate the action of α-synuclein in microglia cells,” he said.—Helen Santoro
References
Research Models Citations
News Citations
Paper Citations
- Lim S, Kim HJ, Kim DK, Lee SJ. Non-cell-autonomous actions of α-synuclein: Implications in glial synucleinopathies. Prog Neurobiol. 2018 Oct;169:158-171. Epub 2018 Jul 3 PubMed.
- Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, Joong Lee S, Masliah E, Hwang D, Lee HJ, Lee SJ. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun. 2013;4:1562. PubMed.
- Kim C, Lee HJ, Masliah E, Lee SJ. Non-cell-autonomous Neurotoxicity of α-synuclein Through Microglial Toll-like Receptor 2. Exp Neurobiol. 2016 Jun;25(3):113-9. Epub 2016 Jun 8 PubMed.
- Moehle MS, Webber PJ, Tse T, Sukar N, Standaert DG, Desilva TM, Cowell RM, West AB. LRRK2 Inhibition Attenuates Microglial Inflammatory Responses. J Neurosci. 2012 Feb 1;32(5):1602-11. PubMed.
- Marker DF, Puccini JM, Mockus TE, Barbieri J, Lu SM, Gelbard HA. LRRK2 kinase inhibition prevents pathological microglial phagocytosis in response to HIV-1 Tat protein. J Neuroinflammation. 2012 Nov 29;9:261. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Kim C, Beilina A, Smith N, Li Y, Kim M, Kumaran R, Kaganovich A, Mamais A, Adame A, Iba M, Kwon S, Lee WJ, Shin SJ, Rissman RA, You S, Lee SJ, Singleton AB, Cookson MR, Masliah E. LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci Transl Med. 2020 Oct 14;12(565) PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
INSERM
The study by Kim and colleagues sheds light on how α-synuclein released from neurons affects a LRRK2-mediated signaling cascade in microglia to promote neuroinflammation. It has been known for some time that LRRK2 is present in the brain in both neurons and microglia. Research on neuronal LRRK2 has revealed effects of LRRK2 on neuromorphology and membrane trafficking, while research on LRRK2 in microglia showed that LRRK2 expression is upregulated upon immune challenge. It has also been shown that α-synuclein-mediated toxicity is dependent on LRRK2, however, the precise mechanisms for these phenomena had not fully been elucidated.
Kim and colleagues first show that exposing microglia to α-synuclein released from neurons activates LRRK2 within these microglia and leads to the release of neurotoxic molecules from these microglia in a LRRK2 kinase activity-dependent manner. Next, transcriptomics analysis of the α-synuclein-exposed microglia allowed the authors to identify the transcription factor NFATc2 as a key player in the microglial LRRK2 cascade. The authors then found that NFATc2 interacted with and was phosphorylated by LRRK2, and that it translocates to the nucleus in a LRRK2-kinase-dependent manner. The authors also found enhanced activation of NFATc2 in postmortem brain tissue from PD and DLB patients and used the Thy1-α-syn mouse to show that microglial activation and motor deficits can be reversed with a LRRK2 kinase inhibitor.
This study is highly significant in that it integrates events from neurons (synuclein release) with the revelation of key elements of a LRRK2-dependent signaling cascade within microglia and how this contributes to generating a toxic environment for neurons. It also shows how one of the proposed therapeutic strategies for PD, i.e., kinase inhibition of LRRK2, blocks the neurotoxic cascade in microglia and is protective against the development of motor symptoms in a transgenic synucleinopathy mouse model of PD.
Interestingly, the authors point to a limitation of LRRK2 inhibition as they observe that the non-motor symptoms as measured in the open-field test were not rescued but rather exacerbated by the inhibitor treatment. This begs further exploration. Would the same observations be made in other synucleinopathy models, such as the nigral synuclein overexpression models or the prion-like models based on CNS injection of aggregated α-synuclein? Can the effects described above on non-motor phenotypes be further characterized in additional tests beyond the open-field test?
Another obvious thought from reading this paper is how these discoveries might open new opportunities for developing new therapeutic strategies and/or improve existing ones. For instance, for the targeting of LRRK2 itself via a kinase inhibitor, are the primary protective effects coming from LRRK2 inhibition in neurons? In microglia? Or does inhibition in both cell types have an additive effect? Answering this question may help determine how therapeutic effectiveness of LRRK2-based therapies can be improved, for instance by targeting the neuron-specific or microglia-specific LRRK2 signaling cascades.
The neurotoxic cascade in microglia revealed by Kim et al. involves extracellular α-synuclein, TLR2, LRRK2, and NFATc2. It will now be interesting to explore which events in this signaling cascade could be targeted to specifically neutralize neurotoxic effects of microglia in synucleinopathy conditions.
Make a Comment
To make a comment you must login or register.