Sans C9ORF72, Microglia Devour More Aβ Plaques. Synapses, Too.
Quick Links
Hexanucleotide expansions within intron 1 of the C9ORF72 gene not only lead to the production of toxic products; they also douse the normal expression of the gene. The consequences of this loss play out dramatically within microglia, according to a study published June 15 in Neuron. Researchers led by Rita Sattler of Barrow Neurological Institute in Phoenix and Robert Baloh of Cedars-Sinai Medical Center in Los Angeles reported that in C9ORF72 knockout mice, microglia rev up expression of interferon genes, accumulate distended lysosomes, and kick synapse destruction into high gear as the mice age. In a mouse model of amyloidosis, C9ORF72 knockouts had fewer, smaller plaques, but also fewer synapses due to overzealous pruning by microglia. In all, the findings pinpoint the inflammatory and degenerative consequences of C9ORF72 loss of function within microglia, and highlight the destructive nature of the interferon response in the brain.
- Microglia lacking C9ORF72 made more interferon.
- They accumulated lysosomes and destroyed synapses.
- 5xFAD mice sans C9 accumulated fewer plaques but lost more synapses, memory.
Hexanucleotide expansions in the C9ORF72 gene lead to amyotrophic lateral sclerosis and/or frontotemporal dementia. Repeat-laden transcripts from the mutated gene weave into RNA foci, while polydipeptide repeats translated from multiple reading frames form protein aggregates. The disruptive expansions also dampen expression of the lysosomal protein, which is involved in autophagy. All told, these toxic gain- and loss of-function mechanisms synergize—churning out a mess of toxic aggregates while also hobbling the cell’s ability to clean them up (Nov 2018 conference news; Zhu et al., 2020).
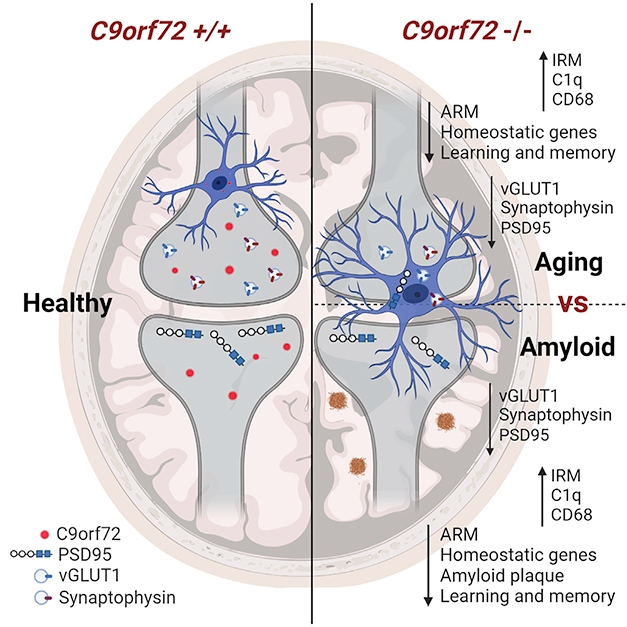
Without C9, Microglia Dine. Healthy microglia that express C9ORF72 leave synapses intact. Without C9, microglia rev up interferon genes with age or during amyloidosis, and overprune synapses. [Courtesy of Lall et al., Neuron, 2021.]
Baloh and colleagues recently reported that in myeloid cells from C9ORF72 knockout mice and from C9 carriers who had ALS/FTD, flagging lysosomal function leads to an accumulation of, among others, the STING protein, a driver of the type I interferon response (McCauley et al., 2020). Interferon escalation and heightened inflammation were known consequences of C9ORF72 knockout (Mar 2016 news).
Could C9 deficiency also mess with microglia? First author Deepti Lall and colleagues addressed this by comparing the transcriptomes of wild-type mouse microglia to those missing one or both copies of C9ORF72. Based on single cell RNA-seq transcriptional profiling, the researchers identified 16 cell clusters, most of which expressed canonical microglial markers P2ry12, TMEM119, and TREM2. Cells in two of these clusters expressed signatures resembling those of activated response microglia (ARM) and interferon response microglia (IRM) that were previously described (Sala Frigerio et al., 2019). ARMs have some similarities with disease associated microglia (DAM) and neurodegenerative microglia (MGnD) described in mouse models of amyloidosis (Apr 2019 news; Jun 2017 news; Sep 2017 news). Lall and colleagues dubbed this cluster activated response microglia (ARM), which was marked by elevated expression of Clec7a, Itgax, ApoE.
IRMs are defined by elevated interferon-stimulated genes, which are akin to those blamed for synapse loss in amyloidosis models and detected in postmortem samples from AD patients (Roy et al., 2020). The proportion of microglia in the ARM and IRM clusters was similar among genotypes. However, the intensity with which cells in each cluster expressed the full ARM or IRM signature differed. In C9 knockouts and in heterozygotes, the IRM signature intensified among IRM cells, while the ARM signature dampened among ARM cells. Notably, these findings meshed with bulk RNA sequencing of microglia, which detected a rise in IRM genes in aged, but not young, C9-deficient mice relative to wild-type.
In keeping with Baloh’s earlier work, the researchers found more STING in C9-deficient microglia, as well as higher expression of downstream, interferon-stimulated genes. C9-less microglia also amassed lysosomes, suggesting a poorly functioning disposal system.
How would these changes influence the way microglia behave? In the motor cortices of year-old C9 knockouts, the researchers found slightly more microglia than in wild-type. They also found more of the complement protein C1q, which is produced by microglia and known to instigate synaptic pruning. Sure enough—more microglia in C9 knockout mice were loaded up with synaptic material than were microglia in wild-type mice, while neurons in C9 knockouts had fewer synapses, fewer dendritic branches, and smaller neurites. Notably, this synaptic dearth was not detected in mice expressing a single copy of C9. In keeping with their sparse synapses, C9 double knockouts had trouble remembering the location of the escape hole in the Barnes maze test of spatial memory.
Are microglial solely to blame for synaptic and behavioral deficits? Recent studies have found that C9-deficiency in neurons can also cause synaptic defects (Xiao et al., 2019; Ho et al., 2019). To eliminate that from the equation, the researchers generated mice lacking C9ORF72 expression only in myeloid cells, including microglia. In these mice, microglia accumulated lysosomes, and synapse numbers were down. This suggested that without C9, microglia transform from neuron protectors to synapse slayers, even when neurons make C9.
Appetite for Amyloid
Microglia are known to overzealously prune synapses in multiple neurodegenerative diseases, including AD. Although C9ORF72 expansions are primarily associated with ALS/FTD, rare cases of C9 expansion carriers with AD or other neurodegenerative diseases have been reported (see Harms et al., 2013; Apr 2014 news). To investigate whether C9 deficiency affects how microglia deal with plaques and synapses in a mouse model of amyloidosis, the researchers generated 5xFAD mice on a C9-deficient background. In 3-month-olds, which are starting to develop amyloid plaques, the scientists spotted no obvious effects of C9 deficiency on plaque deposition. However, by 6 months, 5xFAD mice lacking C9ORF72 had fewer plaques, and those that remained were smaller and more compact than the aggregates in C9-replete 5xFAD mice. In the C9 knockouts, about twice as many microglia crowded around each plaque as in control mice.
What baited more microglia to Aβ plaques in C9 knockout 5xFAD mice? While changes in the microglia themselves could explain it, the researchers also spotted IgG antibodies glommed onto plaques in the C9 knockouts. These antibodies were not present in 5xFAD controls. B cells producing anti-Aβ antibodies were also detected in the spleens and lymph nodes of C9 knockouts, in agreement with the known tendency of C9 knockout mice to churn out autoantibodies. To what extent these anti-Aβ antibodies contributed to microglial plaque clearance remains unclear.
Alas, the stepped-up plaque removal in C9-deficient 5xFAD mice was all for naught, as once again C9 deficiency stoked microglial appetite for synapses. In 4-month-old C9-knockout 5xFAD mice, the researchers found microglia loaded with lysosomes and synaptic proteins, and they detected extensive synapse loss compared to 5xFAD controls. Notably, in C9 knockouts without amyloidosis, these degenerative phenotypes do not emerge until animals are a year old, suggesting that extracellular Aβ accelerated the effects of C9 deficiency. The 5xFAD mice lacking C9 also had more severe spatial memory deficits. In all, the findings suggest that while removing C9ORF72 helps microglia clear plaques, it also triggers synaptic damage that culminates in memory loss.

Synaptic Consumption. In 5xFAD mice (left), microglia (red) contain little synaptic material (PSD95, green). Without C9ORF72, microglia appear loaded with synaptic spoils. [Courtesy of Lall et al., Neuron, 2021.]
“These findings corroborate our previous work showing that IFN-activated microglia are intimately involved in synapse removal, both in wild-type animals and models of Aβ plaque pathology,” commented Wei Cao of Baylor College of Medicine in Houston. “It is interesting to see the increased number of C9ORF72-/- microglia clustered around amyloid plaques; however, it remains to be illuminated if and how the balance between IRMs and ARMs is skewed in 5XFAD by the lack of C9ORF72,” he added. “Better understanding the cross-regulation of different microglial activation states will offer more insights on their functional involvements in pathophysiological processes.”
Sattler told Alzforum that the investigators are using human induced pluripotent stem cell (iPSC)-derived microglia and neurons to understand more about how C9ORF72 loss alters cell functions and interactions. For example, they are investigating whether C9-deficient microglia amass lysosomes because they consume too much debris, and/or because of a slowdown in lysosomal digestion and autophagy. Another lingering question from the study is why only C9ORF72 knockout mice, not heterozygotes, were ravaged by synaptic loss, and what that means for people with ALS or FTD who still carry one functional copy? Sattler noted that it is common for heterozygous mouse models, as in the case of progranulin deficiency, to fall short of replicating the full spectrum of disease in people, who live longer and are different genetically. She believes that studies in cells derived from ALS/FTD IPSCs will more closely model the human disease.—Jessica Shugart
References
News Citations
- It’s ‘And,’ Not ‘Either-Or’: C9ORF72 Mechanisms of Action are Linked
- C9ORF72 Knockout Causes Inflammation, not Neurodegeneration
- Parsing How Alzheimer’s Genetic Risk Works Through Microglia
- Hot DAM: Specific Microglia Engulf Plaques
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
- C9ORF72 Repeats Expand into New Disorders—Cause, or Coincidence?
Paper Citations
- Zhu Q, Jiang J, Gendron TF, McAlonis-Downes M, Jiang L, Taylor A, Diaz Garcia S, Ghosh Dastidar S, Rodriguez MJ, King P, Zhang Y, La Spada AR, Xu H, Petrucelli L, Ravits J, Da Cruz S, Lagier-Tourenne C, Cleveland DW. Reduced C9ORF72 function exacerbates gain of toxicity from ALS/FTD-causing repeat expansion in C9orf72. Nat Neurosci. 2020 May;23(5):615-624. Epub 2020 Apr 13 PubMed.
- McCauley ME, O'Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, Chen S, Lall D, Jin M, Muhammad AK, Bell S, Landeros J, Valencia V, Harms M, Arditi M, Jefferies C, Baloh RH. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020 Sep;585(7823):96-101. Epub 2020 Aug 19 PubMed.
- Sala Frigerio C, Wolfs L, Fattorelli N, Thrupp N, Voytyuk I, Schmidt I, Mancuso R, Chen WT, Woodbury ME, Srivastava G, Möller T, Hudry E, Das S, Saido T, Karran E, Hyman B, Perry VH, Fiers M, De Strooper B. The Major Risk Factors for Alzheimer's Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019 Apr 23;27(4):1293-1306.e6. PubMed.
- Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y, Jankowsky JL, Liu Z, Lee VM, Trojanowski JQ, Ginsberg SD, Butovsky O, Zheng H, Cao W. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020 Apr 1;130(4):1912-1930. PubMed.
- Xiao S, McKeever PM, Lau A, Robertson J. Synaptic localization of C9orf72 regulates post-synaptic glutamate receptor 1 levels. Acta Neuropathol Commun. 2019 Oct 24;7(1):161. PubMed.
- Ho WY, Tai YK, Chang JC, Liang J, Tyan SH, Chen S, Guan JL, Zhou H, Shen HM, Koo E, Ling SC. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy. 2019 May;15(5):827-842. Epub 2019 Jan 28 PubMed.
- Harms M, Benitez BA, Cairns N, Cooper B, Cooper P, Mayo K, Carrell D, Faber K, Williamson J, Bird T, Diaz-Arrastia R, Foroud TM, Boeve BF, Graff-Radford NR, Mayeux R, Chakraverty S, Goate AM, Cruchaga C, . C9orf72 hexanucleotide repeat expansions in clinical Alzheimer disease. JAMA Neurol. 2013 Jun 1;70(6):736-41. PubMed.
Further Reading
Primary Papers
- Lall D, Lorenzini I, Mota TA, Bell S, Mahan TE, Ulrich JD, Davtyan H, Rexach JE, Muhammad AK, Shelest O, Landeros J, Vazquez M, Kim J, Ghaffari L, O'Rourke JG, Geschwind DH, Blurton-Jones M, Holtzman DM, Sattler R, Baloh RH. C9orf72 deficiency promotes microglial-mediated synaptic loss in aging and amyloid accumulation. Neuron. 2021 Jul 21;109(14):2275-2291.e8. Epub 2021 Jun 15 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Baylor College of Medicine
University of Texas Health Science Center at Houston
C9ORF72 repeat expansions associated with neurodegeneration are thought to be detrimental through both accumulation of toxic, gain-of-function protein products and loss of functional C9ORF72 mRNA and protein (LOF). Previous studies from Baloh’s group revealed that C9ORF72 suppresses STING-mediated inflammation in peripheral myeloid cells, which underlies the inflammatory phenotype manifested in C9ORF72-null mice (O'Rourke et al., 2016; McCauley et al., 2020). However, the direct relevance of such LOF phenomenon to CNS neurodegenerative processes was not clear.
Here, Lall et al. focused on the effects of C9ORF72 deficiency in microglia and uncovered enhanced basal inflammation, in particular type I IFN signaling as well as antigen presentation. Single-cell RNA-Seq analysis pinpointed an elevated transcriptional module associated with interferon response microglia (IRM), coupled with diminished activated response microglia (ARM) module. These findings are original and consistent with the crucial function of STING in promoting interferon response and inflammation (Li et al., 2018), and further highlights that microglial activation and subset functionality is critically shaped by innate immune signaling activation.
Using a variety of genetic approaches in vitro and in vivo, Lall et al. revealed an intriguing microglia-intrinsic effect of C9ORF72 deficiency. C9ORF72 -/- microglia, with their heightened IFN signaling, influence aspects of brain homeostasis, such as synaptic density and neuronal morphology, culminating in behavioral deficits.
By analyzing 5XFAD mice crossed to C9ORF72 -/-, the authors found that the deletion severely exacerbated the pathological activities of microglia in the context of AD pathology. These findings corroborate our previous work showing that IFN-activated microglia are intimately involved in synapse removal, both in wild-type animals and in models of Aβ plaque pathology (Roy et al., 2020).
It was interesting to see the increased number of C9ORF72 -/- microglia clustered around amyloid plaques; however, it remains to be determined if and how the balance between IRMs and ARMs is skewed in 5XFAD by the lack of C9ORF72, relative to that observed in wild-type brains. Better understanding of the cross-regulation of different microglial activation states will offer more insights on their functional involvements in pathophysiological processes.
References:
O'Rourke JG, Bogdanik L, Yáñez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J, Kim KJ, Bell S, Harms MB, Miller TM, Dangler CA, Underhill DM, Goodridge HS, Lutz CM, Baloh RH. C9orf72 is required for proper macrophage and microglial function in mice. Science. 2016 Mar 18;351(6279):1324-9. PubMed.
McCauley ME, O'Rourke JG, Yáñez A, Markman JL, Ho R, Wang X, Chen S, Lall D, Jin M, Muhammad AK, Bell S, Landeros J, Valencia V, Harms M, Arditi M, Jefferies C, Baloh RH. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature. 2020 Sep;585(7823):96-101. Epub 2020 Aug 19 PubMed.
Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018 May 7;215(5):1287-1299. Epub 2018 Apr 5 PubMed.
Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y, Jankowsky JL, Liu Z, Lee VM, Trojanowski JQ, Ginsberg SD, Butovsky O, Zheng H, Cao W. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020 Apr 1;130(4):1912-1930. PubMed.
Ann Romney Center for Neurologic Diseases, Brigham and Women's Hospital and Harvard Medical School
This paper from Lall et al. in Neuron is very interesting and consistent with some of our work. C9ORF72 loss of function (due to expanded repeats in human diseases such as ALS/FTD and AD) appears to induce an upregulation of type I Interferon pathways that results in complement deposition and microglia-mediated elimination of complement-tagged synapses. This, in turn, leads to neuronal dysfunction and spatial learning and memory impairment.
Interestingly, C9ORF72 deficiency in 5XFAD mice enhanced IFN response microglia that phagocytosed more Aβ, as well as synapses, leading to lower plaque load, fewer synapses, and enhanced cognitive deficits. Roy and colleagues had demonstrated that the type I Interferon response is upstream of complement signaling and synapse loss (Roy et al., 2020). They found that IFN blockade rescues synapse loss in 5XFAD mice but had no effect on Aβ plaque burden. In our own work, we found that lifelong complement C3-deficiency protected against age-related hippocampal synapse loss and cognitive decline in both WT and APP/PS1 mice (Shi et al., 2015; Shi et al., 2017). However, C3-deficiency also led to increased plaque load in 16-month-old APP/PS1 mice. Interestingly, the microglia in our mice and the amyloid mice with IFN blockade in the Roy paper seemed less activated. This is consistent with the idea that IFN signaling drives a pro-inflammatory signaling cascade that increases complement deposition and synaptic elimination by microglia.
Lall et al. suggest that C9ORF72 may act as a negative regulator of type I Interferon. Thus, when there is C9ORF72 loss of function (due to its expanded repeat), the brakes are released on IFN, which then allows upregulation of complement and its downstream effects. Thus, one would expect less microglial phagocytosis of both synapses and Aβ. Conversely (and consistent with Lall’s results), we observed less microglial phagocytosis of synapses and Aβ, which resulted in sparing of cognitive decline, despite more and larger plaques.
Also, it is very interesting that the loss of C9ORF72 induced anti-amyloid antibodies in the 5XFAD mice. This, too, is likely to involve complement receptor-mediated microglial phagocytosis of amyloid. Lastly, we have now generated global and microglia- or astrocyte-specific C3 inducible conditional knockout mice and are crossing them to APP knock-in mice. It would not be surprising to find cell-autonomous effects of C3 lowering in the brain—time will tell.
References:
Roy ER, Wang B, Wan YW, Chiu G, Cole A, Yin Z, Propson NE, Xu Y, Jankowsky JL, Liu Z, Lee VM, Trojanowski JQ, Ginsberg SD, Butovsky O, Zheng H, Cao W. Type I interferon response drives neuroinflammation and synapse loss in Alzheimer disease. J Clin Invest. 2020 Apr 1;130(4):1912-1930. PubMed.
Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart JC, Caldarone BJ, Stevens B, Lemere CA. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci. 2015 Sep 23;35(38):13029-42. PubMed.
Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017 May 31;9(392) PubMed.
University of Toronto
The article by Lall and colleagues reported important findings on C9ORF72 disease mechanism with significant value for other neurodegenerative disorders. It demonstrated that knockout C9ORF72 mice showed abnormal neuronal morphology, enhanced cortical synaptic loss, and memory deficits. This study revealed the connection between altered microglial function, neurodegeneration, and aging, which has several parallels with our human-based studies.
For instance, we and others demonstrated that increased DNA methylation (DNAm) at the C9ORF72 locus (in response to a repeat-expansion) leads to a downregulation of C9ORF72 expression and correlates with disease onset and duration, indicating that the loss of C9ORF72 could play a role in disease mechanism (Xi et al., 2013; Xi et al., 2015; Gijselinck et al., 2016; McGoldrick et al., 2018). Likely RNA foci and DPRs alone are insufficient to drive neurodegeneration and require additional stressors (e.g., downregulation of C9ORF72). Notably, the highest C9ORF72 levels among CNS tissues was detected in the cerebellum, a region with high abundance of DPRs and RNA foci in C9ORF72 cases but no neurodegeneration (McGoldrick et al., 2018).
The important aspect of Lall et al. is showing the key role of C9ORF72 in antigen presentation and inflammation. Specifically, the loss of C9ORF72 in mice was linked with a significant upregulation of Trem2 and Ctss, encoding cathepsin S that is involved in antigen presentation. Recently, we implicated a 16 Kb region at the 1q21.3 locus as a modifier of ALS age of onset in both C9ORF72 carriers and noncarriers. Importantly, the risk allele is linked with the increased expression of CTSS in cerebellum (Zhang et al., 2021). The role of antigen presenting processes in modulating disease age of onset is also supported by another study, in which we reported that age of onset in a C9ORF72 cohort and general FTD cohort was associated with variants within a 125 Kb region at 6p21.32 locus (Zhang et al., 2018). The risk allele was linked with increased frontal cortex expression of HLA-DRB1 (the major histocompatibility complex class II protein HLA-DR). Notably, HLA-DRB1 is implicated in neurodegenerative diseases as a marker of activated microglia, and important in initiating immune responses by presenting peptides derived from exogenous and endogenous proteins.
The study by Lall et al. also points to the critical connection between neurodegeneration and aging. For instance, it demonstrated that C9ORF72-depleted microglia trigger age-dependent neuronal defects. This brings up another parallel with our human-based studies of ALS and other neurodegenerative diseases. DNA methylation (DNAm) at certain CpG dinucleotides is closely associated with aging (Bergsma and Rogaeva, 2020). The cumulative assessment of DNAm levels at age-related CpGs allow determining DNAm-age reflecting the biological age. DNAm-age acceleration is associated with disease age of onset, duration of survival in C9ORF72-carriers (Zhang et al., 2017), and general cohort of ALS patients (Zhang et al., 2020).
Hence, more studies are needed to understand the role of DNAm-age and immune pathways in neurodegenerative diseases.
References:
Xi Z, Zinman L, Moreno D, Schymick J, Liang Y, Sato C, Zheng Y, Ghani M, Dib S, Keith J, Robertson J, Rogaeva E. Hypermethylation of the CpG Island Near the G4C2 Repeat in ALS with a C9orf72 Expansion. Am J Hum Genet. 2013 May 22; PubMed.
Xi Z, Zhang M, Bruni AC, Maletta RG, Colao R, Fratta P, Polke JM, Sweeney MG, Mudanohwo E, Nacmias B, Sorbi S, Tartaglia MC, Rainero I, Rubino E, Pinessi L, Galimberti D, Surace EI, McGoldrick P, McKeever P, Moreno D, Sato C, Liang Y, Keith J, Zinman L, Robertson J, Rogaeva E. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015 May;129(5):715-27. Epub 2015 Feb 26 PubMed.
Gijselinck I, Van Mossevelde S, van der Zee J, Sieben A, Engelborghs S, De Bleecker J, Ivanoiu A, Deryck O, Edbauer D, Zhang M, Heeman B, Bäumer V, Van den Broeck M, Mattheijssens M, Peeters K, Rogaeva E, De Jonghe P, Cras P, Martin JJ, de Deyn PP, Cruts M, Van Broeckhoven C. The C9orf72 repeat size correlates with onset age of disease, DNA methylation and transcriptional downregulation of the promoter. Mol Psychiatry. 2016 Aug;21(8):1112-24. Epub 2015 Oct 20 PubMed.
McGoldrick P, Zhang M, van Blitterswijk M, Sato C, Moreno D, Xiao S, Zhang AB, McKeever PM, Weichert A, Schneider R, Keith J, Petrucelli L, Rademakers R, Zinman L, Robertson J, Rogaeva E. Unaffected mosaic C9orf72 case: RNA foci, dipeptide proteins, but upregulated C9orf72 expression. Neurology. 2018 Jan 23;90(4):e323-e331. Epub 2017 Dec 27 PubMed.
Zhang M, Xi Z, Saez-Atienzar S, Chia R, Moreno D, Sato C, Montazer Haghighi M, Traynor BJ, Zinman L, Rogaeva E. Combined epigenetic/genetic study identified an ALS age of onset modifier. Acta Neuropathol Commun. 2021 Apr 23;9(1):75. PubMed.
Zhang M, Ferrari R, Tartaglia MC, Keith J, Surace EI, Wolf U, Sato C, Grinberg M, Liang Y, Xi Z, Dupont K, McGoldrick P, Weichert A, McKeever PM, Schneider R, McCorkindale MD, Manzoni C, Rademakers R, Graff-Radford NR, Dickson DW, Parisi JE, Boeve BF, Petersen RC, Miller BL, Seeley WW, van Swieten JC, van Rooij J, Pijnenburg Y, van der Zee J, Van Broeckhoven C, Le Ber I, Van Deerlin V, Suh E, Rohrer JD, Mead S, Graff C, Öijerstedt L, Pickering-Brown S, Rollinson S, Rossi G, Tagliavini F, Brooks WS, Dobson-Stone C, Halliday GM, Hodges JR, Piguet O, Binetti G, Benussi L, Ghidoni R, Nacmias B, Sorbi S, Bruni AC, Galimberti D, Scarpini E, Rainero I, Rubino E, Clarimon J, Lleó A, Ruiz A, Hernández I, Pastor P, Diez-Fairen M, Borroni B, Pasquier F, Deramecourt V, Lebouvier T, Perneczky R, Diehl-Schmid J, Grafman J, Huey ED, Mayeux R, Nalls MA, Hernandez D, Singleton A, Momeni P, Zeng Z, Hardy J, Robertson J, Zinman L, Rogaeva E, International FTD-Genomics Consortium (IFGC). A C6orf10/LOC101929163 locus is associated with age of onset in C9orf72 carriers. Brain. 2018 Oct 1;141(10):2895-2907. PubMed.
Bergsma T, Rogaeva E. DNA Methylation Clocks and Their Predictive Capacity for Aging Phenotypes and Healthspan. Neurosci Insights. 2020;15:2633105520942221. Epub 2020 Jul 21 PubMed.
Zhang M, Tartaglia MC, Moreno D, Sato C, McKeever P, Weichert A, Keith J, Robertson J, Zinman L, Rogaeva E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol. 2017 Aug;134(2):271-279. Epub 2017 Apr 24 PubMed.
Zhang M, McKeever PM, Xi Z, Moreno D, Sato C, Bergsma T, McGoldrick P, Keith J, Robertson J, Zinman L, Rogaeva E. DNA methylation age acceleration is associated with ALS age of onset and survival. Acta Neuropathol. 2020 May;139(5):943-946. Epub 2020 Mar 7 PubMed.
Make a Comment
To make a comment you must login or register.