Ratio of Short to Long Aβ Peptides: Better Handle on Alzheimer's than Aβ42/40?
Quick Links
The Aβ peptides that infamously aggregate in the brains of people with Alzheimer’s disease are products of γ-secretase cleavage of the amyloid precursor protein. Many mutations in the catalytic subunits of the enzyme, as well as its substrate, can cause familial AD, yet the age at which they begin to take their toll varies strikingly from one mutation to the next. Two recent studies assessed what impact mutations have on the full repertoire of Aβ peptides churned out by the secretase. Both found, essentially, that a person's ratio of short to long peptides had a lot to do with when their symptoms started.
- Two studies related the full repertoire of Aβ peptides to age at onset of AD.
- Earlier AAO was tied to low Aβ37/Aβ42, or Aβ (37+38+40)/(42+43).
- These ratios tracked more closely with AAO than did Aβ42/40.
One, published March 12 in Alzheimer’s and Dementia and led by Dennis Selkoe of Brigham and Women’s Hospital in Boston, reported that lower ratios of Aβ37 to Aβ42 correlated with earlier age at onset (AAO) in people with familial AD, and also with greater Aβ burden and more cognitive impairment in people with sporadic disease. The Aβ37/42 ratio outperformed the Aβ42/40 ratio, which has become a staple biomarker for AD and has entered clinical practice. The other study was led by Lucia Chávez-Gutiérrez at KU Leuven in Belgium and published April 1 in Molecular Psychiatry. It found that ratios of short to long peptides, particularly the Aβ (37+38+40)/(42+43) ratio, best correlated with AAO in people carrying known ADAD mutations, and even predicted AAO in variants with unclear pathogenicity.
Notably, the Aβ42/40 ratio did not consistently track with AAO in either study. Overall, the findings suggest that Aβ peptide ratios that reflect the hobbling of γ-secretase processivity could serve as more sensitive biomarkers for disease onset and severity.
“Taken together, these studies support the critical importance of comprehensive evaluation of Aβ peptide profiles in deciphering AD pathogenesis,” noted Chávez-Gutiérrez.
Gamma-secretase handles its substrate—the C99 fragment of APP—via two consecutive cleavage events: ε endoprotease cleavage produces the long, membrane-anchored peptides Aβ48 or Aβ49, off which γ-cleavage then slices three to four amino acids at a time until the remaining peptide is released from the plasma membrane. In this manner, two product lines of Aβ peptides are released: Aβ 49→46→43→40→37 and Aβ 48→45→42→39, 38 or 37.
Familial AD mutations in PSEN1 destabilize the γ-secretase complex, hobbling its processive function, and leading to relative overproduction of the longer, aggregation-prone peptides Aβ42 and Aβ43 at the expense of the shorter peptides (Szaruga et al., 2017). Most studies in the field have focused on Aβ42, yet have come to different conclusions about how the Aβ42/40 ratio reflects disease severity.
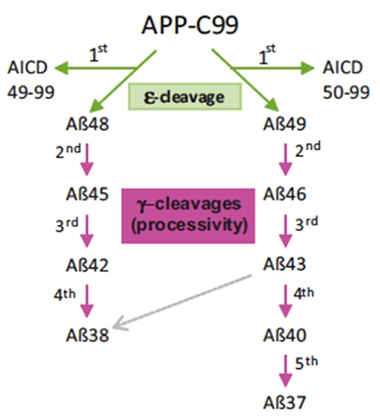
Assembly Line. Gamma secretase first cleaves the APP-C99 substrate into either Aβ48 or Aβ49 peptides via ε-cleavage. Next, it trims these substrates to produce an array of Aβ peptides. [Courtesy of Petit et al., Molecular Psychiatry, 2022.]
Might accounting for the full Aβ peptide repertoire be more useful? In Selkoe’s group, first author Lei Liu and colleagues used immunoassays they had developed to measure all six Aβ peptides produced by cells expressing different PSEN1 variants. In a nutshell, they found that, compared to Aβ42/40, the Aβ37/42 ratio correlated more robustly and consistently with the AAO associated with each of more than 100 PSEN1 mutations.
This ratio of short-to-long peptides also proved informative in sporadic AD. In either brain tissue or in CSF, Aβ37/42 distinguished between people with low and high Aβ burden, and also separated people who were cognitively normal from those who had been impaired.
In Chávez-Gutiérrez’s study, first author Dieter Petit and colleagues measured the spectrum of Aβ peptides released by cells expressing any of 25 different familial AD mutations in PSEN1. Using an unbiased statistical approach, they found that the Aβ (37+38+40)/(42+43) ratio correlated with the AAO associated with each mutation.
When the scientists used this ratio to estimate the AAO of PSEN1 mutations whose pathogenicity was unclear, such as those identified in only one person with AD, the estimated AAO fell within six years of the reported AAO for most mutations. In some exceptions where the estimate was further off the mark, other genetic influences, such as ApoE genotype, could account for the discrepancy. The findings suggest not only that changes in the Aβ (37+38+40)/(42+43) ratio—i.e., γ-secretase processive activity—track closely with AAO, but also that additional genetic and/or environmental factors tweak this relationship.
“Overall, the study further confirms the crucial role diverse Aβ species play in AD pathology, and that Aβ profiles of several Aβ species (not only Aβ40 and Aβ42) best predict AAO,” wrote Rebecca Gabriele, Selina Wray, and Charlie Arber of University College London in a joint comment to Alzforum. “Moreover, AAO is strongly linked to longer, more aggregation-prone Aβ species, suggesting that shifting Aβ profiles toward shorter and less amyloidogenic peptides could be a valid therapeutic approach.”
More generally, the shorter Aβ species are getting a second wind (Jan 2022 news; Feb 2022 news). One γ-secretase modulator is poised to enter clinical trials (Rynearson et al., 2021); alas, most pharma companies are believed to have ended their efforts to develop such compounds.—Jessica Shugart
References
News Citations
- Does More Aβ38 Mean Less Cognitive Decline in Alzheimer’s?
- “Frustrated Oligomers” Slow Aggregation of Aβ42
Paper Citations
- Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
- Rynearson KD, Ponnusamy M, Prikhodko O, Xie Y, Zhang C, Nguyen P, Hug B, Sawa M, Becker A, Spencer B, Florio J, Mante M, Salehi B, Arias C, Galasko D, Head BP, Johnson G, Lin JH, Duddy SK, Rissman RA, Mobley WC, Thinakaran G, Tanzi RE, Wagner SL. Preclinical validation of a potent γ-secretase modulator for Alzheimer's disease prevention. J Exp Med. 2021 Apr 5;218(4) PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Liu L, Lauro BM, He A, Lee H, Bhattarai S, Wolfe MS, Bennett DA, Karch CM, Young-Pearse T, Dominantly Inherited Alzheimer Network (DIAN), Selkoe DJ. Identification of the Aβ37/42 peptide ratio in CSF as an improved Aβ biomarker for Alzheimer's disease. Alzheimers Dement. 2022 Mar 12; PubMed.
- Petit D, Fernández SG, Zoltowska KM, Enzlein T, Ryan NS, O'Connor A, Szaruga M, Hill E, Vandenberghe R, Fox NC, Chávez-Gutiérrez L. Aβ profiles generated by Alzheimer's disease causing PSEN1 variants determine the pathogenicity of the mutation and predict age at disease onset. Mol Psychiatry. 2022 Jun;27(6):2821-2832. Epub 2022 Apr 1 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Lund University
I found the associations between the Aβ(37+38+40)/Aβ(42+43) ratio and the age at onset of symptoms in PSEN mutations carriers reported by the Chavez-Guttierez group, and between the Aβ37/42 ratio and pathogenicity and clinical diagnosis as reported by the Selkoe lab, very convincing.
To me, these results provide further evidence that modulation of the γ-secretase protease, shifting Aβ generation toward shorter peptides, is a very interesting therapeutic approach for both familial and non-familial forms of AD. However, clinical trials evaluating such γ-secretase modulators should likely be performed during the early, asymptomatic stages of the disease to have a reasonable chance to substantially diminish the accumulation of toxic Aβ aggregates.
University College London
UCL Queen Square Institute of Neurology
Institute of Neurology, University College London
The paper by Petit and colleagues emphasizes an aspect of AD pathology that is increasingly gaining attention: the importance of Aβ species other than Aβ40 and Aβ42 in AD onset and progression.
FAD-associated mutations in PSEN1 destabilize the interaction between the γ-secretase complex and APP/Aβn substrate, promoting the formation of longer aggregation-prone Aβ peptides (Arber et al., 2019; Szaruga et al., 2021). Yet the age of onset (AAO) in PSEN1 mutant carriers varies incredibly. Why? This article reveals that the (37 + 38 + 40) / (42 + 43) ratio not only predicts AAO but does so more accurately than the canonical Aβ42/40 ratio, underlying the importance of additional Aβ species.
In line with this, the paper by Liu and colleagues suggests that the Aβ37/42 ratio in the CSF, rather than the Aβ42/40 ratio, is a better biomarker for AD pathology for familial and sporadic cases. Moreover, a mixture of all Aβ37, Aβ38, and Aβ40 has a greater inhibitory effect on Aβ42 aggregation compared to each single peptide in vitro (Braun et al., 2022). Importantly, these findings further distinguish longer, more aggregation-prone Aβ42 and Aβ43 peptides from shorter species in pathogenesis and clinical onset in AD.
The fact that the profiles of Aβ species predict AAO suggests that we could use such a profile to understand if novel PSEN1 mutations are pathogenic or not. This would have a fundamental impact in the clinical setting. Of note, AD is a complex disease where environmental and other genetic factors can also influence AAO. As discussed by Petit et al., APOE2 can delay AAO by eight years.
We eagerly anticipate follow-up studies that will determine the similarities/differences when it comes to employing these ratios to estimate AAO of APP mutations—e.g., mutations around γ-secretase versus β-secretase cleavage sites. We also look forward to seeing these ratios in action in clinical settings, where the healthy allele may lead to more complex profiles that the mutation in isolation.
Overall, Petit et al. further confirm the crucial role diverse Aβ species play in AD pathology, and that Aβ profiles of several Aβ species (not only Aβ40 and Aβ42) best predict AAO. Moreover, AAO is strongly linked to longer, more aggregation-prone Aβ species, suggesting that shifting Aβ profiles toward shorter and less-amyloidogenic peptides could be a valid therapeutic approach.
References:
Arber C, Villegas-Llerena C, Toombs J, Pocock JM, Ryan NS, Fox NC, Zetterberg H, Hardy J, Wray S. Amyloid precursor protein processing in human neurons with an allelic series of the PSEN1 intron 4 deletion mutation and total presenilin-1 knockout. Brain Commun. 2019;1(1):fcz024. Epub 2019 Oct 14 PubMed.
Braun GA, Dear AJ, Sanagavarapu K, Zetterberg H, Linse S. Amyloid-β peptide 37, 38 and 40 individually and cooperatively inhibit amyloid-β 42 aggregation. Chem Sci. 2022 Feb 23;13(8):2423-2439. Epub 2022 Feb 7 PubMed.
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell. 2021 Apr 15;184(8):2257-2258. PubMed.
K.U.Leuven and V.I.B.
I read this extensive and comprehensive report by Lei Liu et al. with great interest. Indeed, this and our study present complementary data and insights, as well as tools for the quantitative assessment of familial and sporadic AD.
In the context of familial AD: Liu et al. focused on the analysis of single Aβ ratios versus AAO, while we looked at other Aβ peptide combinations based on γ-secretase mechanisms. We asked what peptide combination is the best predictor of AAO. We also performed a data-driven approach testing Aβ-AAO relationships; this analysis provided complementary insights into differential roles and contributions of Aβ peptides to pathology.
Liu et al. identified the Aβ37/42 ratio as the best benchmark for differentiating FAD variants from the WT PSEN1. In line with their findings, our studies support the notion that the relationship between short and longer Aβ peptides plays a critical role in AD pathogenesis. In addition, our analyses show that the Aβ(37+38+40)/(42+43) ratio has predictive value in the assessment of AAO.
Liu et al. also investigated the Aβ37/42 ratio in the context of the most common, sporadic AD. Interestingly, their analyses of brain samples revealed that the Aβ37/42 relationship better differentiates AD from non-demented, low-amyloid-pathology cases, relative to the canonical Aβ42/40 ratio. Furthermore, the assessment of Aβ peptide levels in CSF samples from AD, mild cognitive impairment (MCI) and control cases revealed that the Aβ37/42 ratio outperforms the Aβ42/40 ratio in distinguishing control versus disease (AD or MCI) cases.
That both familial and sporadic samples showed similar alterations in the Aβ37/42 ratio is an intriguing observation. In this regard, it is worth noting previous studies associating changes in Aβ37 production levels, relative to Aβ42, with amyloid plaque deposition and cognitive trajectory (Lagomarsino et al., 2021). Why do changes in short versus longer Aβ peptides occur in SAD? This definitely warrants further investigation.
Taken together, these studies support the critical importance of comprehensive evaluation of Aβ peptide profiles in deciphering AD pathogenesis.
References:
Lagomarsino VN, Pearse RV 2nd, Liu L, Hsieh YC, Fernandez MA, Vinton EA, Paull D, Felsky D, Tasaki S, Gaiteri C, Vardarajan B, Lee H, Muratore CR, Benoit CR, Chou V, Fancher SB, He A, Merchant JP, Duong DM, Martinez H, Zhou M, Bah F, Vicent MA, Stricker JM, Xu J, Dammer EB, Levey AI, Chibnik LB, Menon V, Seyfried NT, De Jager PL, Noggle S, Selkoe DJ, Bennett DA, Young-Pearse TL. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron. 2021 Nov 3;109(21):3402-3420.e9. Epub 2021 Sep 1 PubMed.
Washington University
Outside of Aβ42 and Aβ40, few species of Aβ are typically studied. Liu and colleagues performed a variety of experiments to characterize less-commonly-described forms of Aβ: They examined Aβ profiles produced by cell lines expressing familial AD mutations, Aβ profiles in human brain tissue, and Aβ profiles in human CSF.
In the last set of experiments, they reported that CSF Aβ37/Aβ42 outperformed CSF Aβ42/Aβ40 in distinguishing MCI and/or AD cases from cognitively normal controls (ROC AUC of 0.96 versus 0.87). However, the cohort they used was very small (38 cognitively normal, 25 MCI, and 16 AD dementia), and it was not clear that the AD diagnosis was confirmed with AD biomarkers (e.g. amyloid PET). In fact, a couple of MCI cases had relatively high CSF Aβ42/Aβ40, and since CSF Aβ42/Aβ40 is a highly sensitive biomarker of amyloidosis, it seems possible that the cohort included some non-AD causes of MCI, complicating the interpretation.
As the authors noted, low CSF Aβ42/Aβ40 identifies individuals with brain amyloidosis, including cognitively normal individuals with brain amyloidosis, and prediction of clinical diagnosis based on CSF Aβ42/Aβ40 is often not very robust. Also, CSF Aβ42/Aβ40 plateaus after individuals develop brain amyloidosis (the so-called L-shape), and may not distinguish well between clinical diagnoses/stages. If the results of this study are replicated and Aβ37/Aβ42 is better correlated with clinical diagnosis/stage, it may suggest that Aβ37/Aβ42 is changing later in the AD course, allowing for better prediction of clinical diagnosis/stage.
Both Liu et al. and Petit et al. examined Aβ profiles associated with many different familial AD mutations. Interestingly, the Aβ profiles produced by cells expressing these familial AD mutations were associated with the typical age at symptom onset for the mutation. Liu et al. found strong associations of both Aβ42/Aβ40 and Aβ37/Aβ42 with the age at symptom onset; Petit et al. found a strong association of (Aβ37 + Aβ38 + Aβ40)/(Aβ42 + Aβ43) and a moderate association of Aβ42/Aβ40 with the age at symptom onset.
Overall, this suggests that ratios combining multiple short-to-long species of Aβ could potentially be better biomarkers of AD than Aβ42/Aβ40. However, it will be important to evaluate this hypothesis in sporadic AD.
AlzeCure Pharma
The results presented in the two papers by Liu et al. and Petit et al., respectively, are highly interesting. They suggest that shorter Aβ species could play an important role in predicting age at onset (AAO), but potentially could also be important from a therapeutic perspective as the results suggest that shifting Aβ profiles toward shorter and less-amyloidogenic peptides could be a valid approach. These data, as well as results from Cullen et al. earlier this year, show that higher CSF Aβ38 levels are associated with less cognitive decline and lower risk of developing AD, give further credit to the hypothesis that a lower ratio of shorter to longer Aβ peptides could play a role in amyloid toxicity and pathogenicity. In light of these findings, it was also interesting to read the recent paper by Braun et al., where they investigated the aggregation of various Aβ species and were able to demonstrate that Aβ37, 38, and 40 individually and cooperatively inhibit Aβ42 aggregation.
γ-Secretase modulators, such as AlzeCure's compounds ACD679 and ACD680, represent a promising class of Aβ42-lowering anti-amyloidogenic compounds for the treatment of AD. GSMs exhibit several key features that make them suitable for the treatment in early AD, especially in view of the results discussed above: 1) They reduce amyloidogenic Aβ42 production while stimulating the formation of Aβ37 and 38, and 2) they modulate but do not affect total γ-secretase activity, a property that is of central importance from a safety perspective. As such, GSMs modulate the formation of secreted Aβ, while sparing the γ-secretase-mediated processing event resulting in the release of the cytoplasmic APP intracellular domain making GSMs an interesting Aβ-targeting therapy in early AD. A new wave of AD therapeutics will likely include GSMs and it will be highly interesting to see the outcome of these clinical studies.
References:
Cullen N, Janelidze S, Palmqvist S, Stomrud E, Mattsson-Carlgren N, Hansson O, Alzheimer’s Disease Neuroimaging Initiative. Association of CSF Aβ38 Levels With Risk of Alzheimer Disease-Related Decline. Neurology. 2022 Mar 1;98(9):e958-e967. Epub 2021 Dec 22 PubMed.
Braun GA, Dear AJ, Sanagavarapu K, Zetterberg H, Linse S. Amyloid-β peptide 37, 38 and 40 individually and cooperatively inhibit amyloid-β 42 aggregation. Chem Sci. 2022 Feb 23;13(8):2423-2439. Epub 2022 Feb 7 PubMed.
Make a Comment
To make a comment you must login or register.