PET Firms Up Amyloid Cascade: Plaques, Inflammation, Tangles
Quick Links
In the Alzheimer’s cascade hypothesis, plaques unleash tangles; alas, where neuroinflammation fits in has been hazy. Now, the first study to combine imaging of microglial activation with amyloid and tau PET in the human brain places neuroinflammation squarely in between the two. In the August 26 Nature Medicine, researchers led by Tharick Pascoal, now at the University of Pittsburgh School of Medicine, and Pedro Rosa-Neto at McGill University in Montreal, report PET findings from 108 adults who range from cognitively healthy to AD dementia.
- The regional pattern of microglial activation in AD brain mirrors Braak staging.
- In the presence of plaque, it parallels and predicts tangle spread.
- The data suggest that amyloid and microglia together unleash tangles.
Across this cohort, the regional distribution of microglial activation mirrored Braak staging, and correlated with tangle load. Moreover, the extent of microglial activation predicted the spread of tangles into later Braak regions, suggesting it drove this pathology. Notably, the relationship between neuroinflammation and tangles only occurred in the presence of amyloid plaques, and all three pathologies were required for cognitive decline.
"Amyloid potentiates microglial activation to drive tau propagation in the brain,” Pascoal told Alzforum. "The data suggest neuroinflammation should be included in biological definitions of AD.” Some of these data were previously presented at the 2020 Human Amyloid Imaging conference (Feb 2020 conference news).

Plaque + Microglia = Tangles
In people with a high plaque load (red scale), the extent of microglial activation (X axis) correlated with how widespread tangles were (Y axis). Without plaque (gray), this relationship disappeared. [Courtesy of Pascoal et al., Nature Medicine.]
Others were enthusiastic. "This is a very compelling study, and certainly advances our understanding of the cross talk between microglial activation, amyloid, and tau burden in the clinical context," Kiran Bhaskar at the University of New Mexico, Albuquerque, wrote to Alzforum (full comment below). Marc Diamond at the University of Texas Southwestern in Dallas said it would spur further research. “This type of correlation is a powerful inspiration to labs such as mine to test whether there are certain inflammatory signals that might influence tau propagation,” he wrote (comment below).
Neuroinflammation Linked to Tangles
PET imaging studies have consistently shown that as plaques spread into the cortex, tangles break out of the medial temporal lobe to rampage across the brain, attacking cognition as they go (Mar 2016 news; Aug 2016 news; Feb 2018 news). But the mechanistic connection between the pathologies remained mysterious. The medial temporal lobe contains little amyloid, making a direct interaction unlikely.
Animal and in vitro studies hinted that microglia might be the missing link. Michael Heneka at the German Center for Neurodegenerative Diseases in Bonn reported that in mice, activation of the NLRP3 inflammasome in microglia caused the cells to spew cytokines that triggered tau phosphorylation in neurons (Nov 2019 news). Work by Sarah Hopp at Massachusetts General Hospital in Charlestown suggested a different microglial mechanism. She found that microglia isolated from AD brain contained tau seeds, which the cells released into the culture medium. The data implied that microglia phagocytose aggregated neuronal tau present in aging brain, then try but fail to digest it, and instead end up strewing it across the brain (Hopp et al., 2018).
Pascoal and colleagues directly examined microglia in living humans via PET. They used PBR28, a tracer that binds the mitochondrial protein TSPO. TSPO is predominantly expressed by activated microglia, but also sometimes by activated astrocytes and endothelium. Unfortunately, a common polymorphism in TSPO interferes with binding, leading to a low PET signal in many people (Apr 2012 news). To get around this problem, the authors studied only people with no copies of this polymorphism, which amounted to about half of those they genotyped. Pascoal noted that neuroinflammation does not vary between low- and high-affinity TSPO binders, so he believes the findings from this study will generalize to the broader population. Bhaskar praised the decision to exclude low binders, because this enabled the authors to collect more-consistent data in which variation in the PET signal reflected disease stage rather than genetic differences.
Participants came from McGill’s Translational Biomarkers of Aging and Dementia (TRIAD) cohort. After screening out people with neurological conditions other than AD, the researchers had 130 participants, 22 of whom were healthy young controls with an average age of 23. The older participants had an average age of 72; 64 of them were cognitively healthy, 28 had mild cognitive impairment, and 16 had AD dementia. All participants underwent cognitive testing, MRI, and PET scans at baseline, and the older group had a follow-up tau PET scan one year later.
What did PBR28 show? Uptake of the tracer associated with high cerebrospinal fluid sTREM2, another marker of microglial activation that is elevated in AD. Moreover, brain regions in cognitively healthy older participants that bound PBR28 matched those with high TREM2 expression in postmortem brain samples from the Allen Human Brain Atlas, suggesting the PBR28 signal reflects microglial activation. The strength of the global PBR28 signal roughly correlated with the global amyloid signal, as seen by the tracer AZD4694, and with overall tangle load, measured with MK6240.
In terms of its regional pattern, PBR28 uptake rose with age, particularly in the precuneus, posterior cingulate, lateral temporal, and inferior parietal cortex. However, the pattern changed with disease, lighting up more regions in MCI and AD brain. Like tangles, microglial activation progressed from a Braak stage I pattern in cognitively healthy participants up to Braak stage VI in people at more advanced stages of AD.
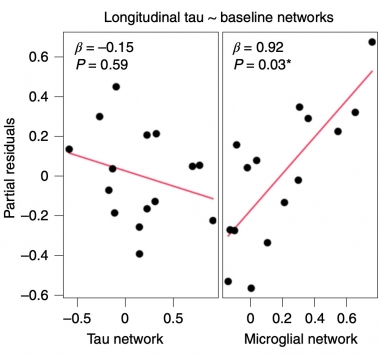
Microglia Make Tangles Spread
The baseline tau PET network does not correlate with longitudinal change in tangles (left), but the strength of the microglial network at baseline does (right). [Courtesy of Pascoal et al., Nature Medicine.]
PBR28 uptake also correlated with the change in tau PET over one year. A regression analysis of the baseline PBR28 and MK6240 regional networks found that the microglial network strongly predicted future tangle spread, with a correlation coefficient of 0.92, while the baseline tau PET network did not (see image at left). In more evidence of a causal role for microglial activation, the PBR28 signal at baseline in the transentorhinal cortex, i.e. in Braak stage I, foreshadowed tangle accumulation in later Braak stage regions on the follow-up scan.
"The exciting observations from Pascoal and colleagues that tau pathology follows changes in microglia activation align with over two decades of cellular and mouse research suggesting that microglia pro-inflammatory activation drives tau pathology," Hopp noted (full comment below).
Amyloid Sets the Stage
Where does amyloid fit in? Both high plaque load and microglial activation were required for tangles to occur. Against a background of high cortical plaque load, the more widespread a person's activated microglia were, the more widespread their tangles. For example, people with a high global amyloid burden who bound PBR28 in the transentorhinal cortex had tangles in the hippocampal region, while people who bound PBR28 in Braak stage IV regions had widespread neocortical tangles. In the absence of amyloid, this relationship between inflammation and tangles vanished (see image at top of story).
To put the icing on the PET cake, Pascoal found that all three pathologies acted together to precipitate cognitive decline. The presence of any single pathology had no consistent effect on cognition, but having all three together hiked a person's risk 25-fold.
The Nuances of Microglial Activation
The study raises several questions, in particular how microglia promote tangle spread, and what type of activation does this. TSPO binding is a blunt instrument that cannot distinguish nuanced microglial phenotypes. Some types of microglial activation, at the right stage of disease, may even help ameliorate pathology. For example, mouse studies suggest that TREM2 activation early in amyloidosis helps corral plaques and neutralize toxicity (May 2016 news; Apr 2020 news; Jun 2020 news).
David Holtzman at Washington University in St. Louis noted that mouse studies from his group have found that microglial activation curbs tau seeding, rather than promotes it (Leyns et al., 2019; Jul 2019 conference news; Gratuze et al., 2021). "We always need to remember that correlation does not equal causation," he wrote to Alzforum (full comment below). Could it be that the active microglia coincide with tangles because they are trying to corral or eliminate them, as they do with plaques (Sep 2019 news; Apr 2021 news)?
Rosa-Neto will tackle the causation question by manipulating microglial reactivity, for example by modulating TREM2, and measuring how that affects tau pathology in animal models. He also wants to pin down the type of microglial activation in AD brain by examining fluid biomarkers of inflammation. In this PET study, the PBR28 signal correlated with numerous CSF inflammatory markers, including fractalkine and its receptor CX3CR1, TGF-α, CSF-1, and IL-8. Some of these markers correlated more closely with the PBR28 signal than did sTREM2. Bhaskar’s research has shown CX3CR1 strongly affects tau pathology in mouse models, suggesting a key role for this receptor.
"CSF biomarkers may better capture a snapshot of the neuroinflammatory phenotype [than does PBR28]," Hopp agreed.
Rosa-Neto will continue to follow this cohort, adding longitudinal PBR28 and amyloid scans as well as additional tau scans to learn more about when the various pathologies appear and change in relation to one another. However, he believes existing data already suggest that to be effective, anti-inflammatory therapy would have to be started early in AD, before tangles break out of the medial temporal lobe. Recent findings that fluid markers of inflammation change very early in preclinical disease strengthen this idea (Aug 2021 conference news). Past anti-inflammatory trials, for example one of minocycline in mild AD dementia, may have begun too late in disease to do any good, Rosa-Neto speculated (Nov 2019 news).—Madolyn Bowman Rogers
References
News Citations
- How Much Amyloid Will Kick Off Tangles, and Decline?
- Tau PET Aligns Spread of Pathology with Alzheimer’s Staging
- Brain Imaging Suggests Aβ Unleashes the Deadly Side of Tau
- Imaging Clinches Causal Connections between Aβ, Tau, Circuitry, and Cognition
- Microglia Inflammasome Stokes Tau Phosphorylation, Tangles
- Imaging Inflammation: Can Glial PET Tracers Make a Mark?
- Barrier Function: TREM2 Helps Microglia to Compact Amyloid Plaques
- With TREM2, Timing Is Everything
- In Mice, Activating TREM2 Tempers Plaque Toxicity, not Load
- TREM2, Microglia Dampen Dangerous Liaisons Between Aβ and Tau
- Are Microglia Plaque Factories?
- Microglia Build Plaques to Protect the Brain
- Mirror, Mirror on the Wall, Who’s the Earliest of Them All?
- Minocycline Does Not Work in Mild Alzheimer’s Disease
Paper Citations
- Hopp SC, Lin Y, Oakley D, Roe AD, DeVos SL, Hanlon D, Hyman BT. The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer's disease. J Neuroinflammation. 2018 Sep 18;15(1):269. PubMed.
- Leyns CE, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VM, Ulrich JD, Holtzman DM. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci. 2019 Aug;22(8):1217-1222. Epub 2019 Jun 24 PubMed.
- Gratuze M, Chen Y, Parhizkar S, Jain N, Strickland MR, Serrano JR, Colonna M, Ulrich JD, Holtzman DM. Activated microglia mitigate Aβ-associated tau seeding and spreading. J Exp Med. 2021 Aug 2;218(8) Epub 2021 Jun 8 PubMed.
Further Reading
Papers
- Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, Savard M, Lussier FZ, Tissot C, Karikari TK, Ottoy J, Mathotaarachchi S, Stevenson J, Massarweh G, Schöll M, de Leon MJ, Soucy JP, Edison P, Blennow K, Zetterberg H, Gauthier S, Rosa-Neto P. Microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021 Sep;27(9):1592-1599. Epub 2021 Aug 26 PubMed. Correction.
News
- Individualized Tau PET Model Outperforms Predictive Power of Braak Staging
- Simulating Tangle Spread Along Axons, Scientists Predict Tau PET Patterns
- New at Tau2020: PET Detects First Traces of Tangles in Rhinal Cortex
- Tau PET Scans Turn Positive When Amyloid Does; Symptoms Follow
- Serial PET Nails It: Preclinical AD Means Amyloid, Tau, then Cognitive Decline
- Longitudinal Tau PET Links Aβ to Subsequent Rise in Cortical Tau
- In Familial Alzheimer’s, Tau Creeps into Cortex as Symptoms Show
- Analysis of PET Scans Suggests Link Between Amyloid and Tau
- Do Temporal Lobe Tangles and Cortical Plaques Together Bring on Alzheimer’s?
Primary Papers
- Pascoal TA, Benedet AL, Ashton NJ, Kang MS, Therriault J, Chamoun M, Savard M, Lussier FZ, Tissot C, Karikari TK, Ottoy J, Mathotaarachchi S, Stevenson J, Massarweh G, Schöll M, de Leon MJ, Soucy JP, Edison P, Blennow K, Zetterberg H, Gauthier S, Rosa-Neto P. Microglial activation and tau propagate jointly across Braak stages. Nat Med. 2021 Sep;27(9):1592-1599. Epub 2021 Aug 26 PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
This is a very nice manuscript that reports associations between amyloid, microglial activation, and tau pathology with correlations suggesting that amyloid-related microglial activation correlates with both tau pathology and with cognitive decline.
One of the things we always need to remember is that correlation does not equal causation. Data from our lab suggests that in the presence of amyloid deposition, microglial activation is linked with less, not more, amyloid-related tau seeding and spreading (Leyns et al., 2019). In addition, removing microglia in an amyloid model also increases tau seeding and spreading (Gratuze et al., 2021).
With no TREM2, with R47H TREM2, or with no microglia, all of which decrease microglial activation, there is actually significantly greater, not less, tau seeding and spreading. Obviously, mouse model data may not mimic what occurs in humans; however, in this case, I think it offers direct experimental evidence suggesting the opposite of what the authors claim in this manuscript regarding the role of microglial activation in tau spreading.
References:
Leyns CE, Gratuze M, Narasimhan S, Jain N, Koscal LJ, Jiang H, Manis M, Colonna M, Lee VM, Ulrich JD, Holtzman DM. TREM2 function impedes tau seeding in neuritic plaques. Nat Neurosci. 2019 Aug;22(8):1217-1222. Epub 2019 Jun 24 PubMed.
Gratuze M, Chen Y, Parhizkar S, Jain N, Strickland MR, Serrano JR, Colonna M, Ulrich JD, Holtzman DM. Activated microglia mitigate Aβ-associated tau seeding and spreading. J Exp Med. 2021 Aug 2;218(8) Epub 2021 Jun 8 PubMed.
UT Health Science Center San Antonio
The exciting observations from Pascoal and colleagues that tau pathology follows changes in microglia activation aligns with over two decades of cellular and mouse research suggesting that microglia pro-inflammatory activation drives tau pathology. Dr. W. Sue Griffin was perhaps one of the earliest proponents of a “cytokine cycle,” whereby microglial activation drives tau hyperphosphorylation, an important precursor for tau aggregation. Later research from Dr. Kiran Bhaskar and colleagues in mice further supported the idea that reactive microglia could initiate tau hyperphosphorylation and spreading.
I was particularly excited by the observation that several other CSF inflammatory proteins correlated with PBR28 SUVR, most notably soluble CX3CL1, a ligand for the microglia receptor CX3CR1, which has been crucially linked to microglial propagation of tau pathology by Bhaskar and colleagues, as well as more recently by Bolós et al.
My research, as well as research from Dr. Tsuneya Ikezu, demonstrates that microglia directly spread tau pathology by releasing tau seeds that are capable of inducing tau aggregation in recipient cells. Importantly, the observation that tau is not strongly associated with Aβ without the presence of microglia activation supports the idea that microglia potentiate tau spread, either directly (via tau release) or indirectly (via inflammatory signaling cascades).
While this study by Pascoal and colleagues cannot pinpoint whether microglia inflammatory cascades, or microglia release of tau seeds, explains their observation that tau pathology follows changes in the microglia activation network, it offers critical support for observations that microglia influence tau propagation in animal and cellular models that recapitulate processes that occur in the human AD brain.
It is important to note that microglia, including activated microglia, can also play important roles in restricting the propagation of tau pathology. My research, and that of others, has shown that microglia are capable of internalizing and degrading tau seeds, thus reducing propagation to other cells. Activation of microglia via inflammagens, such as lipopolysaccharide, can enhance tau uptake and degradation by microglia.
Further, Gratuze et al. recently reported that, in the context of Aβ, knockout of TREM2, which suppressed microglia activation, enhanced tau spread, as did microglia ablation. The beneficial roles of microglia in restriction of tau propagation are not entirely consistent, however, since Clayton et al. recently observed that microglia depletion reduced tau spread in the context of Aβ.
Transcriptomics studies may help resolve this discrepancy, since they reveal that microglia display different activation phenotypes at different stages of AD, whereby early microglia activation may restrict pathology while late microglia activation may exacerbate it. Therefore, it is important to consider both the beneficial and detrimental aspects of microglia activation when using ligands such as PBR28, which capture an incomplete characterization of the microglia phenotype. Importantly, the authors correlated PBR28 with CSF biomarkers that may better capture a snapshot of the neuroinflammatory phenotype at the time of data collection; Pascoal et al.’s data point toward a detrimental role for microglial pro-inflammatory activation in tau propagation in patients with AD.
Overall, these results strengthen the connection between microglia activation and tau pathology by providing key clinical data that support previous experimental evidence from cellular and animal models of tau pathology. This research highlights the importance of tracking microglia activation as new therapeutics progress in clinical trials.
University of Texas, Southwestern Medical Center
This is a fascinating study in that it shows the strong correlation of microglial activation (whatever that means) with the progression of tau pathology. The authors posit a causal connection, which is interesting but not clearly suggested by the data. It seems equally plausible that tau propagation could feed back to influence microglial activation.
Certainly, this type of correlation is a powerful inspiration to labs such as mine to test whether there are certain inflammatory signals that might influence tau propagation.
Karolinska Institutet
This very interesting study focuses on the possible importance microglial activation may have for the spreading of tau tangles in the brain and their co-localizing in a Braak pattern. A total of 130 subjects across a spectrum, including 22 young and 64 older cognitively unimpaired individuals, 28 subjects with mild cognitive impairment, and 16 patients with Alzheimer´s disease, underwent PET investigations with three tracers: [11C]-PBR28 PET (microglia activation), [18F]-AZD4694 (amyloid), [18F]-MK6240 (tau). An increased binding of [11C]-PBR28 was observed in older subjects with low Aβ and tau tracer binding. Microglial activation together with amyloid and tau accumulation turned out to be a strong predictor of cognitive decline. The study clearly illustrates the important role of early microglial activation for development and progression of AD pathology and clinical symptoms, an observation that might be crucial when designing new disease modifying drugs for AD.
In a recent study, high [11C]-PBR28 binding in brain was measured in healthy elderly who had low levels of amyloid in the brain and were considered to be amyloid PIB negative (Toppala et al. 2021). A biphasic increase in microglial activation has been suggested during AD progression, with an early protective microglial response followed by a late, more pro-inflammatory effect (Leng and Edison, 2021). Both microglia and astrocytes have, in contrast to Aβ and tau tangles, important physiological functions in the brain, both during development and in adult and older states, and cross-talk seems to exist between glia cells and neurons. Tau has been reported to accumulate in astrocytes, which made Alzforum question “Does astrocytes tau cause dementia?” (Nov 2020 news). It would be very interesting to perform studies with simultaneous PET imaging of microglial activation and astrogliosis during progression to AD from normal aging.
Using [11C]-deprenyl PET, our research group has reported high astrogliosis in autosomal-dominant AD mutation carriers 15 years prior to their estimated onset of clinical symptoms (Rodriguez-Vietiez et al., 2016) and with divergent longitudinal changes in astrogliosis compared to amyloid PET. We also found significant positive associations between longitudinal astrogliosis ([11C]-deprenyl ) and 18F-FDG PET (Carter et al., 2019), as well as cortical microstructural changes that correlate with astrogliosis in presymptomatic AD mutation carriers (Vilaplana et al., 2020). Are microglia and astrocytes both activated in the early presymptomatic stages of AD? And do both synergistically drive tau propagation?
Further studies of glia and tau imaging are ongoing in AD. New PET tracers, [11C]-BU99008 and [18F]-SMBT-1, have recently been developed for imaging astrogliosis, and both appear promising (Calsolaro et al., 2021; Kumar et al., 2021; Harada et al., 2021). New microglia PET tracers are urgently needed to circumvent the problem of the TSPO polymorphism that affects PBR28 binding. In the present study, Pascoal and colleagues had to genotype 503 individuals to identify 130 with high-affinity TSPO binding sites, something that would be difficult and time-consuming in a clinical trial design.
References:
Toppala S, Ekblad LL, Tuisku J, Helin S, Johansson JJ, Laine H, Löyttyniemi E, Marjamäki P, Blennow K, Zetterberg H, Jula A, Viitanen M, Rinne JO. Association of Early β-Amyloid Accumulation and Neuroinflammation Measured With [11C]PBR28 in Elderly Individuals Without Dementia. Neurology. 2021 Mar 23;96(12):e1608-e1619. Epub 2021 Jan 29 PubMed.
Leng F, Edison P. Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here?. Nat Rev Neurol. 2021 Mar;17(3):157-172. Epub 2020 Dec 14 PubMed.
Carter SF, Chiotis K, Nordberg A, Rodriguez-Vieitez E. Longitudinal association between astrocyte function and glucose metabolism in autosomal dominant Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2019 Feb;46(2):348-356. Epub 2018 Dec 4 PubMed.
Vilaplana E, Rodriguez-Vieitez E, Ferreira D, Montal V, Almkvist O, Wall A, Lleó A, Westman E, Graff C, Fortea J, Nordberg A. Cortical microstructural correlates of astrocytosis in autosomal-dominant Alzheimer disease. Neurology. 2020 May 12;94(19):e2026-e2036. Epub 2020 Apr 14 PubMed.
Calsolaro V, Matthews PM, Donat CK, Livingston NR, Femminella GD, Guedes SS, Myers J, Fan Z, Tyacke RJ, Venkataraman AV, Perneczky R, Gunn R, Rabiner EA, Gentleman S, Parker CA, Murphy PS, Wren PB, Hinz R, Sastre M, Nutt DJ, Edison P. Astrocyte reactivity with late-onset cognitive impairment assessed in vivo using 11C-BU99008 PET and its relationship with amyloid load. Mol Psychiatry. 2021 Jul 15; PubMed.
Kumar A, Koistinen NA, Malarte ML, Nennesmo I, Ingelsson M, Ghetti B, Lemoine L, Nordberg A. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer's disease brain. Mol Psychiatry. 2021 Apr 23; PubMed.
Harada R, Hayakawa Y, Ezura M, Lerdsirisuk P, Du Y, Ishikawa Y, Iwata R, Shidahara M, Ishiki A, Kikuchi A, Arai H, Kudo Y, Yanai K, Furumoto S, Okamura N. 18F-SMBT-1: A Selective and Reversible PET Tracer for Monoamine Oxidase-B Imaging. J Nucl Med. 2021 Feb;62(2):253-258. Epub 2020 Jul 9 PubMed.
Cyceron
In this study, Pascoal et al. showed from PET imaging for microglial activation, amyloid, and tau pathologies in about 130 individuals, that i) the baseline microglial activation, but not tangle, network strongly predicted longitudinal tau propagation; ii) global Aβ promoted this process, as has been seen in prior studies; iii) the synergistic effects of the three processes associated with dementia, leading to worse MMSE scores. The authors proposed that the interaction between regional activated microglia and global amyloid deposition catalyzes tau propagation.
This is a particularly interesting study in the fact that it brings new—and possibly key—elements to our understanding of the pathophysiological mechanisms of the disease; or at least it suggests, in an elegant study, a new possible path to explore.
Let’s take a few steps back in how we conceptualized those mechanisms. The advent of molecular neuroimaging techniques allowing us to visualize in vivo how AD pathologies per se, namely amyloid deposition and tau, have “shaken” the amyloid cascade hypothesis (see e.g., Knopman et al., 2012, and the following discussion by Vishnu, 2013; Chételat 2013, 2013). We then progressively evolved from a conception of a disease with a single unitary pathological event, where Aβ/amyloid was blamed for everything, to a more multi-determinate concept of the disease, acknowledging multiple possible pathological pathways. The idea that Aβ and tau could occur partly independently, at least in the medial temporal lobe, started to emerge, i.e., we started to see amyloid as not being the only possible actor—and tau/neurofibrillary tangles as a possibly partly independent pathological process. As I wrote in 2013, “We are entering an era in which the unitary view of AD as a disease with a single sequential pathological pathway—with Aβ considered as the only initial and causal event—is likely to be progressively replaced by a more complex picture in which AD is considered as a multiparameter pathology that is subtended by several partly independent pathological processes. … For example, Aβ, tau and possibly other pathologies may be partly independent of one another, each under the influence of both independent and common risk factors, and able to interact with each other, thereby promoting the AD neuropathological cascade.” (Chételat 2013).
Altogether, there is more and more acknowledgement in the field that other pathological elements might also intervene. Notably, the process of tau propagation and related atrophy is still a mystery and a topic of great interest that is stimulating ingenious multimodal neuroimaging studies. Such studies have shown that connectivity from a starting point, i.e., the “epicenter,” is involved, consistent with the neuronal spread hypothesis (Raj et al., 2015; Yang et al., 2019; Mutlu et al., 2017), through an active rather than a passive diffusion process, and that functionally strong and spatially short connections increase the likelihood of tau seeding and spread (Franzmeier et al., 2020).
First it was thought that the propagation from the medial temporal lobe to the neocortex depended on amyloid; but studies then showed that this spreading process, and the network specificity of tau, was not dependent on the presence of amyloid (i.e., it could occur without Aβ), but that the presence of amyloid (i.e., global amyloid instead of regional) might promote the process (Franzmeier et al., 2019; Sepulcre et al., 2016). However, the regional topography of the epicenter still appeared insufficient to explain the specificity of tau propagation. The spatial and even microstructural (cellular) specificity of where the pathology starts turns out to be relevant to explain the specific topography of tau spreading (Bejanin et al., 2017; de Flores et al., 2020; Duyckaerts et al., 2013). Importantly, other mechanisms were thought to intervene as well to trigger this propagation and to explain its topographical specificity.
There was evidence for a role of microglial activation in this process from animal studies but not in human. The study by Pascoal et al. offers evidence in human and goes a step forward in proposing a model with a specific role for activated microglial as a new active actor of the pathological process of AD, joining the ranks of, and interacting with, amyloid and tau pathologies.
These findings are notably important for development of new therapeutic strategies aiming to contain disease progression. Of course more work is needed i) to confirm these results (e.g., with better PET tracers and or solving some methodological issues related with this PET measure of microglial activation; with larger/independent cohorts, with longer-term follow-up), and ii) to further understand what is responsible for microglial activation, and what is responsible for the topography of microglial activation and spread. Indeed, if the specific topography of tau spreading is explained, at least partly, by the microglial activation topography, the latter remains to be understood.
In sum, these results fit well with the more and more accepted view that AD is not due to a single pathological actor (responsible for all other events/manifestations as in the amyloid cascade) but the result of the complex interaction between different actors. We started to accept the view that amyloid pathology may not be responsible for all the damage; we progressively started to think that Aβ and tau (and maybe other pathological elements) might progressively and partly independently accumulate, until they interact and trigger a cascading event leading to accelerated neurodegeneration and dementia. In this study by Pascoal et al., a role is assigned to microglia activation as a major actor in this process, specifying the function this actor might play, and thereby opening the avenue to the exploration of this new possible pathway.
References:
Bejanin A, Desgranges B, La Joie R, Landeau B, Perrotin A, Mézenge F, Belliard S, de La Sayette V, Eustache F, Chételat G. Distinct white matter injury associated with medial temporal lobe atrophy in Alzheimer's versus semantic dementia. Hum Brain Mapp. 2017 Apr;38(4):1791-1800. Epub 2016 Dec 16 PubMed.
Chételat G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat Rev Neurol. 2013 Mar;9(3):123-4. PubMed.
Chételat G. Reply: The amyloid cascade is not the only pathway to AD. Nat Rev Neurol. 2013 Jun;9(6):356. Epub 2013 May 14 PubMed.
Duyckaerts C. Neurodegenerative lesions: Seeding and spreading. Rev Neurol (Paris). 2013 Oct;169(10):825-33. PubMed.
de Flores R, Wisse LE, Das SR, Xie L, McMillan CT, Trojanowski JQ, Robinson JL, Grossman M, Lee E, Irwin DJ, Yushkevich PA, Wolk DA. Contribution of mixed pathology to medial temporal lobe atrophy in Alzheimer's disease. Alzheimers Dement. 2020 Jun;16(6):843-852. Epub 2020 Apr 22 PubMed.
Franzmeier N, Rubinski A, Neitzel J, Kim Y, Damm A, Na DL, Kim HJ, Lyoo CH, Cho H, Finsterwalder S, Duering M, Seo SW, Ewers M, Alzheimer’s Disease Neuroimaging Initiative. Functional connectivity associated with tau levels in ageing, Alzheimer's, and small vessel disease. Brain. 2019 Apr 1;142(4):1093-1107. PubMed.
Franzmeier N, Neitzel J, Rubinski A, Smith R, Strandberg O, Ossenkoppele R, Hansson O, Ewers M, Alzheimer’s Disease Neuroimaging Initiative (ADNI). Functional brain architecture is associated with the rate of tau accumulation in Alzheimer's disease. Nat Commun. 2020 Jan 17;11(1):347. PubMed.
Knopman DS, Jack CR, Wiste HJ, Weigand SD, Vemuri P, Lowe VJ, Kantarci K, Gunter JL, Senjem ML, Mielke MM, Roberts RO, Boeve BF, Petersen RC. Brain injury biomarkers are not dependent on β-amyloid in normal elderly. Ann Neurol. 2012 Nov 23; PubMed.
Mutlu J, Landeau B, Gaubert M, de La Sayette V, Desgranges B, Chételat G. Distinct influence of specific versus global connectivity on the different Alzheimer's disease biomarkers. Brain. 2017 Dec 1;140(12):3317-3328. PubMed.
Raj A, LoCastro E, Kuceyeski A, Tosun D, Relkin N, Weiner M, for the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Network Diffusion Model of Progression Predicts Longitudinal Patterns of Atrophy and Metabolism in Alzheimer's Disease. Cell Rep. 2015 Jan 14; PubMed.
Sepulcre J, Schultz AP, Sabuncu M, Gomez-Isla T, Chhatwal J, Becker A, Sperling R, Johnson KA. In Vivo Tau, Amyloid, and Gray Matter Profiles in the Aging Brain. J Neurosci. 2016 Jul 13;36(28):7364-74. PubMed.
Vishnu VY. Can tauopathy shake the amyloid cascade hypothesis?. Nat Rev Neurol. 2013 Jun;9(6):356. Epub 2013 May 14 PubMed.
Yang F, Roy Chowdhury S, Jacobs HI, Johnson KA, Dutta J. A longitudinal model for tau aggregation in Alzheimer's disease based on structural connectivity. Inf Process Med Imaging. 2019;11492:384-393. Epub 2019 May 22 PubMed.
University of New Mexico
This very interesting paper provides direct evidence for the role of microglial activation and amyloidosis in driving tau propagation, especially in live human brain. This study's careful design recruited only high responders of TSPO, which is very important. The statistical analysis and other methods are also appropriate to draw their conclusions on positive correlations between the spread of microglial activation and tau. Profiling of sTREM2 and other immune regulators in the CSF, and correlating them with microglial and tau spread via Braak staging brain regions, are also strengths of this study.
In view of recent evidence on the crosstalk between microglial activation and tau pathology pertinent to dementia, this study fits well with the tau-microglia interactions in non-AD tauopathies originally observed by Girard’s (Gerhard et al., 2006) and Dixon’s (Ishizawa and Dickson, 2001) groups, and it complements recent reports of the role of microglia on tau spread via exosomes or otherwise.
A few other studies, including our own, complement this study. We found that the deletion of microglia-specific fractalkine receptor (CX3CR1) was sufficient to accelerate tau pathology in hTau mice, a preclinical animal model of tauopathy (Bhaskar et al., 2010) and that adoptive transfer of these reactive CX3CR1-deficient microglia was sufficient to drive tau pathology in recipient mice brains (Maphis et al., 2015). This link (microglia driving tau pathology) was also observed by Li Gan’s group in an amyloid-based model (Cho et al., 2011). In view of these studies, I think it is important to note that Rosa-Neto and colleagues have observed significant upregulation of CX3CL1 in the CSF, which also significantly correlated with [11C]PRB28PET. This observation is compelling and suggests neuron-microglia cross-talk via CX3CL1-CX3CR1 is important and warrants further investigation in human subjects/samples.
On a related note, since Aβ fuels microglia-driven tau spread corresponding to Braak staging, it is important to see if microglia-driven tau spread also occurs in non-AD tauopathies (such as PSP, CBD, PiD etc.) via PET imaging, especially since Virginia Lee’s group has first reported that microglial activation can precede tau pathology in their PS19 mouse model of pure tauopathy (Yoshiyama et al., 2007).
Finally, on the same note, and given that we have a new FDA-approved drug for AD, it would be interesting to see if patients taking aducanumab who had reduced levels of Aβ burden would show uncoupling of [11C]PBR28 and [18F]MK-6240 uptake in Braak staging brain regions.
Nonetheless, this compelling study certainly advances our understanding of the cross talk between microglial activation, amyloid, and tau burden in the clinical context.
References:
Gerhard A, Trender-Gerhard I, Turkheimer F, Quinn NP, Bhatia KP, Brooks DJ. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in progressive supranuclear palsy. Mov Disord. 2006 Jan;21(1):89-93. PubMed.
Ishizawa K, Dickson DW. Microglial activation parallels system degeneration in progressive supranuclear palsy and corticobasal degeneration. J Neuropathol Exp Neurol. 2001 Jun;60(6):647-57. PubMed.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010 Oct 6;68(1):19-31. PubMed.
Maphis N, Xu G, Kokiko-Cochran ON, Jiang S, Cardona A, Ransohoff RM, Lamb BT, Bhaskar K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015 Jun;138(Pt 6):1738-55. Epub 2015 Mar 31 PubMed.
Cho SH, Sun B, Zhou Y, Kauppinen TM, Halabisky B, Wes P, Ransohoff RM, Gan L. CX3CR1 protein signaling modulates microglial activation and protects against plaque-independent cognitive deficits in a mouse model of Alzheimer disease. J Biol Chem. 2011 Sep 16;286(37):32713-22. Epub 2011 Jul 19 PubMed.
Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007 Feb 1;53(3):337-51. PubMed.
University of Southampton
University of Southampton, Faculty of Medicine
An important point about this impressive study is that it is in living humans and so can give information about the time course of evolution of AD pathology, otherwise difficult to access. The findings fit well with a hypothesis for interactions of Aβ, microglia and tau in causing Alzheimer’s disease that we recently proposed, which puts some human neuropathological detail into the sequence, as follows:
Step i: Many elderly and middle-aged people have diffuse cortical Aβ plaques, i.e., plaques lacking microglia, dystrophic neurites and p-tau. They are cognitively normal, indicating that the diffuse Aβ plaques by themselves do not cause dementia.
Step ii: Microglia recognize the presence of Aβ plaques and respond by migrating toward the plaques, abandoning their normal homeostatic functions, acquiring activated morphology, secreting neurotoxic substances and initiating the complement cascade. Most genetic factors increasing risk for, but not causing, AD are associated with microglia and operate at this step. As an end product of the complement cascade, membrane attack complex forms pores, evolved to killed invading micro-organisms, in nearby neuronal processes; the resultant metabolic dysregulation disrupts the neuritic cytoskeleton and neuronal transport, causing the characteristic irregularly thickened and tortuous dystrophic neurites of the neuritic plaque which accumulate APP as a reflection of the disturbed transport (APP+/p-tau- dystrophic neurites).
Step iii: The associated neuritic dysfunction facilitates spread of aggregated p-tau, which has already begun to accumulate in limbic structures as part of the aging process (Primary Age-Related Tauopathy), to the neocortex, first appearing in the plaque-associated dystrophic neurites (APP+/p-tau + dystrophic neurites).
Step iv: P-tau spreads within neurons from the dystrophic neurites to the neuronal somata, resulting in neurofibrillary tangles. Subsequently, p-tau spreads by cell-cell transfer throughout the neocortex, resulting in progressive neurodegeneration and dementia.
Microglial functions are altered in three ways: (i) loss of normal homeostatic functions that support neuronal and synaptic health; (ii) a toxic reaction to Aβ plaques resulting in bystander damage to neuronal processes, facilitating p-tau accumulation, and (iii) clearance of neuronal debris. APOE ε4, the main genetic risk factor for AD, has multiple actions including: increased Aβ accumulation, enhanced microglial reactivity and impaired clearance of neuronal debris.
This hypothesis implies different therapeutic targets are appropriate for different steps of AD pathogenesis:
Step i – Primary prevention, disrupt Aβ aggregation, e.g. active Aβ immunotherapy.
Step ii – Secondary prevention, modulate microglial activity.
Steps iii/iv – Established AD, target p-tau.
References:
Boche D, Nicoll JA. Invited Review - Understanding cause and effect in Alzheimer's pathophysiology: Implications for clinical trials. Neuropathol Appl Neurobiol. 2020 Dec;46(7):623-640. Epub 2020 Jul 25 PubMed.
McGill University Faculty of Medicine
I respectfully disagree with Drs. Bhaskar ("driving tau propagation"), Chetelat ("strongly predicted"), Nordberg ("important role of early microglial activation for development and progression of AD pathology"), and Hopp ("tau pathology follows changes in microglia activation").
I urge consideration instead of Drs. Holtzman and Diamond's reminder that correlation does not mean causation. Indeed, it does not imply directionality. Relevant here: All trials of anti-inflammatory treatments in AD have yielded null results, or worse. Be it prednisone (Aisen et al., 2000), hydroxychloroquine (Van Gool et al., 2001), naproxen (Aisen et al., 2000; ADAPT Research Group et al., 2007; Meyer et al., 2019), rofecoxib (Aisen et al., 2000; Thal et al., 2005), or celecoxib (ADAPT Research Group et al., 2007) the uniform result has been no benefit.
In the instance of celecoxib, rofecoxib, and naproxen, or all NSAIDs as a class (Aisen et al., 2000; ADAPT Research Group et al., 2007; Meyer et al., 2019; Thal et al., 2005; Breitner et al., 2009; Sonnen et al., 2010), there have been significantly worse outcomes when symptomatic cases of AD or MCI (most, presumably, due to AD) have been exposed to anti-inflammatory treatments. Could these consistent results not suggest that the glial response observed by Pascoal, Rosa-Neto and others to accompany tau tangle formation is an adaptive or defensive response (Meyer et al., 2018)?
Other forms of analysis will be needed to determine directionality (and surely, causality) as an explanation for this association.
References:
Aisen PS, Davis KL, Berg JD, Schafer K, Campbell K, Thomas RG, Weiner MF, Farlow MR, Sano M, Grundman M, Thal LJ. A randomized controlled trial of prednisone in Alzheimer's disease. Alzheimer's Disease Cooperative Study. Neurology. 2000 Feb 8;54(3):588-93. PubMed.
Van Gool WA, Weinstein HC, Scheltens P, Walstra GJ, Scheltens PK. Effect of hydroxychloroquine on progression of dementia in early Alzheimer's disease: an 18-month randomised, double-blind, placebo-controlled study. Lancet. 2001 Aug 11;358(9280):455-60. PubMed.
ADAPT Research Group, Lyketsos CG, Breitner JC, Green RC, Martin BK, Meinert C, Piantadosi S, Sabbagh M. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007 May 22;68(21):1800-8. Epub 2007 Apr 25 PubMed.
Meyer PF, Tremblay-Mercier J, Leoutsakos J, Madjar C, Lafaille-Maignan MÉ, Savard M, Rosa-Neto P, Poirier J, Etienne P, Breitner J, PREVENT-AD Research Group. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. 2019 Apr 30;92(18):e2070-e2080. Epub 2019 Apr 5 PubMed.
Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, Reines SA, . A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology. 2005 Jun;30(6):1204-15. PubMed.
Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009 Jun 2;72(22):1899-905. PubMed.
Sonnen JA, Larson EB, Walker RL, Haneuse S, Crane PK, Gray SL, Breitner JC, Montine TJ. Nonsteroidal anti-inflammatory drugs are associated with increased neuritic plaques. Neurology. 2010 Sep 28;75(13):1203-10. PubMed.
Meyer PF, Savard M, Poirier J, Labonté A, Rosa-Neto P, Weitz TM, Town T, Breitner J, Alzheimer’s Disease Neuroimaging Initiative, PREVENT-AD Research Group. Bi-directional Association of Cerebrospinal Fluid Immune Markers with Stage of Alzheimer's Disease Pathogenesis. J Alzheimers Dis. 2018;63(2):577-590. PubMed.
McGill University
We would like to express our gratitude for the very insightful comments from colleagues on our publication. These discussions are extremely important for designing studies to answer the several questions raised by our results.
Regarding previous reports suggesting a protective effect of microglia activation in AD, it is crucial to consider the multidimensional approach to neuroinflammation. For example, while the PET ligand PBR28 reveals over-expression of the 18kD translocator protein (TSPO) in the mitochondria of brain cells, mainly in activated microglia, other neuroinflammation markers might potentially indicate a different facet of the so-called "microglial activation." In fact, the interpretation of biomarker results should consider that brain inflammatory responses encompass multiple cell types, cellular compartments, and downstream pathways; hence it is expected that their associations with AD pathophysiology might differ among biomarkers (ligands, antibodies, etc.).
One should be cautious about the interpretation of clinical trials targeting neuroinflammation pathways in AD. In fact, design and methodological issues in some of these anti-inflammation trials limit inferences regarding disease pathophysiology (Meyer et al., 2019; Meyer et. al., 2019). Therefore, the absence of efficacy in these trials does not necessarily refute the role of neuroinflammation in the diathesis of AD.
We are excited to continue our multimodal longitudinal studies and hope our findings stimulate studies focusing on innovative therapeutic strategies in AD.
References:
Meyer PF, Labonté A, Rosa-Neto P, Poirier J, Breitner JC, PREVENT‐AD Research Group. No apparent effect of naproxen on CSF markers of innate immune activation. Ann Clin Transl Neurol. 2019 Jun;6(6):1127-1133. Epub 2019 Jun 6 PubMed.
Meyer PF, Tremblay-Mercier J, Leoutsakos J, Madjar C, Lafaille-Maignan MÉ, Savard M, Rosa-Neto P, Poirier J, Etienne P, Breitner J, PREVENT-AD Research Group. INTREPAD: A randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology. 2019 Apr 30;92(18):e2070-e2080. Epub 2019 Apr 5 PubMed.
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
Biomedical Center (BMC), Metabolic Biochemistry, Faculty of Medicine, Ludwig-Maximilians University and German Centre for Neurodegenerative Diseases (DZNE), Munich, Germany
As already pointed out by David Holtzmann, cross-sectional correlations between biomarkers in an observational study do not imply causation. Moreover, the claim that microglial activation together with amyloid accumulation is a driver of tau spreading should be discussed in the context of our extensive knowledge on protective functions of TREM2 (summarized by Lewcock et al., 2020). In that context, we should also remember that disease-associated TREM2 variants result in a reduced function of TREM2, an observation that is difficult to reconcile with the authors’ interpretation.
Moreover, in the presence of amyloid plaques, loss of TREM2 has been shown to facilitate tau accumulation, brain atrophy and amyloid pathology, thus, TREM2 activation is protective (Lee et al., 2021).
One can of course argue that these are data from animal models, but there is also plenty of evidence in humans that TREM2 may in fact exert protective functions. For example, there is an association between higher sTREM2 at baseline and a slower cognitive decline, hippocampal shrinkage and amyloid accumulation in sporadic AD in a symptomatic phase (Deming et al., 2019; Ewers et al., 2019; Ewers et al., 2020).
Moreover, our recent findings in the longitudinal observational DIAN study also show that a higher longitudinal sTREM2 increase in the pre-symptomatic AD phase up to 20 years before symptom onset highly correlates with a slower disease progression as measured by longitudinal cognitive scores, molecular and structural neuroimaging (Haass et al., 2021). We also observed that patients with lower longitudinal sTREM2 increase had a higher increase in CSF p-tau related to the amyloid-PET signal than the patients with a higher longitudinal sTREM2 increase (Haass et al., 2021), supporting the results by Lee et al. 2021 from a clinical perspective.
The conclusions and discussion of the observations made by Pascoal et al. would have benefited from a balanced discussion in the context of the current knowledge on TREM2 function in model systems and humans.
References:
Deming Y, Filipello F, Cignarella F, Cantoni C, Hsu S, Mikesell R, Li Z, Del-Aguila JL, Dube U, Farias FG, Bradley J, Budde J, Ibanez L, Fernandez MV, Blennow K, Zetterberg H, Heslegrave A, Johansson PM, Svensson J, Nellgård B, Lleo A, Alcolea D, Clarimon J, Rami L, Molinuevo JL, Suárez-Calvet M, Morenas-Rodríguez E, Kleinberger G, Ewers M, Harari O, Haass C, Brett TJ, Benitez BA, Karch CM, Piccio L, Cruchaga C. The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer's disease risk. Sci Transl Med. 2019 Aug 14;11(505) PubMed.
Ewers M, Franzmeier N, Suárez-Calvet M, Morenas-Rodriguez E, Caballero MA, Kleinberger G, Piccio L, Cruchaga C, Deming Y, Dichgans M, Trojanowski JQ, Shaw LM, Weiner MW, Haass C, Alzheimer’s Disease Neuroimaging Initiative. Increased soluble TREM2 in cerebrospinal fluid is associated with reduced cognitive and clinical decline in Alzheimer's disease. Sci Transl Med. 2019 Aug 28;11(507) PubMed.
Ewers M, Biechele G, Suárez-Calvet M, Sacher C, Blume T, Morenas-Rodriguez E, Deming Y, Piccio L, Cruchaga C, Kleinberger G, Shaw L, Trojanowski JQ, Herms J, Dichgans M, Alzheimer's Disease Neuroimaging Initiative (ADNI), Brendel M, Haass C, Franzmeier N. Higher CSF sTREM2 and microglia activation are associated with slower rates of beta-amyloid accumulation. EMBO Mol Med. 2020 Sep 7;12(9):e12308. Epub 2020 Aug 10 PubMed.
Lee SH, Meilandt WJ, Xie L, Gandham VD, Ngu H, Barck KH, Rezzonico MG, Imperio J, Lalehzadeh G, Huntley MA, Stark KL, Foreman O, Carano RA, Friedman BA, Sheng M, Easton A, Bohlen CJ, Hansen DV. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron. 2021 Apr 21;109(8):1283-1301.e6. Epub 2021 Mar 5 PubMed.
Lewcock JW, Schlepckow K, Di Paolo G, Tahirovic S, Monroe KM, Haass C. Emerging Microglia Biology Defines Novel Therapeutic Approaches for Alzheimer's Disease. Neuron. 2020 Dec 9;108(5):801-821. Epub 2020 Oct 22 PubMed.
Haass C, Morenas-Rodriguez E, Li Y, Franzmeier N, Xiong C, Suárez-Calvet M, Fagan A, Schultz S, Gordon B, Benzinger T, Hassenstab J, McDade E, Feederle R, Karch C, Schlepckow K, Morris J, Kleinberger G, Nellgård B, Vöglein J, Blennow K, Zetterberg H, Ewers M, Jucker M, Levin J, Bateman R. Longitudinal increase of CSF soluble TREM2 is driven by early aggregation of Aβ42 and associates with slower amyloid deposition and clinical decline in autosomal-dominant Alzheimer’s disease. Research Square. Version 1 Posted 31 Jul, 2021. Research Square.
University of Pittsburgh
We would like to thank everyone for the great discussion. Our work showed a link between microglial activation brain circuits measured with a PET proxy of TSPO availability and tau tangles propagation. We did show a positive association between brain TSPO availability and CSF TREM2, as we showed with 55 other inflammation-related proteins. Some of these other proteins showed a better association with TSPO results than TREM2 and, likely, several other untested targets would also represent better TSPO PET outcomes.
Although the positive association with TREM2 (and other proteins) supported PET TSPO as a marker of neuroinflammation/microglial activation, more elaborate conclusions about the role of TREM2 in tau progression should be addressed in studies designed to understand this pathway, which was not the case in our work.
Make a Comment
To make a comment you must login or register.