Novel Exome Screen Points to Tubulin as ALS Gene
Quick Links
A new kind of genetic analysis has identified a gene that likely contributes to risk for amyotrophic lateral sclerosis. As reported in the October 22 Neuron, geneticists scanning the exomes of ALS cases and controls found that people with the disease were far more likely to contain variants in the tubulin, alpha 4A gene (TUBA4A). Experts who spoke with Alzforum said that although the evidence is strong, an independent study pointing to TUBA4A would be needed to confirm the association. Senior author John Landers of the University of Massachusetts (UMass) Medical School in Worcester also urged caution, calling TUBA4A a risk factor but not necessarily a causative gene. TUBA4A joins several other cytoskeleton-related genes that have been implicated in neurodegeneration, bolstering the theory that cellular struts and beams keep neurons healthy.
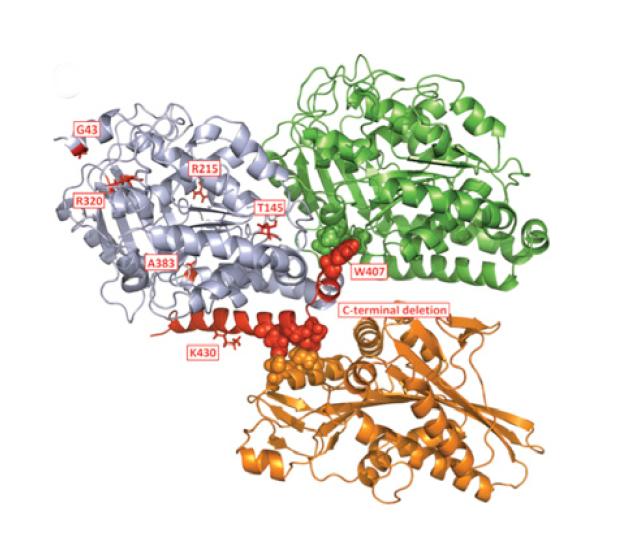
Altered Interface: Alpha-tubulin (light blue) interacts with the β isoform (green) and also kinesin (orange). The red residues indicate those affected by human mutations or truncations found in ALS cases. [Image courtesy of Neuron, Smith et al.]
Traditionally, one way geneticists found new disease genes was by analyzing large kindreds that had been affected. As recently as March of this year, researchers in Italy and the United States used this approach to find mutations in Matrin 3, a DNA- and RNA-binding protein, that associate with ALS (Johnson et al., 2014). However, geneticists have already studied most of the large U.S. and European families with ALS. “We had to come up with an alternative strategy,” said Landers.
Landers began an international collaboration with co-senior authors Vincenzo Silani of the University of Milan Medical School and Christopher Shaw of King’s College London. Their study is one of the first to use a method called exome-wide rare variant analysis (Panoutsopoulou et al., 2013; Stitziel et al., 2011). In this technique, researchers compare the exomes of people with the disease with those of healthy controls. Genes more frequently altered in people with ALS are deemed likely to influence disease risk. “This does not say that any one of those variants, in particular, is causative,” cautioned Landers. He said some variants may turn out to be harmless. The researchers and Neuron have produced a video abstract that explains the strategy.
The first step for joint first authors Bradley Smith, Athina Gkazi, and Simon Topp of King’s College, Nicola Ticozzi of the University of Milan Medical School, and Claudia Fallini of UMass Medical School was to read the exomes of 363 people who had inherited ALS but had no mutations in any of the known ALS genes. To find genes that varied more in ALS, they compared those sequences to 4,300 control exomes from a National Institutes of Health database. To avoid benign changes in the genetic code, the authors limited their analysis to variants predicted to alter protein structure or function by the bioinformatics tool PolyPhen-2.
The researchers built in a positive control by including exome data from people with mutations in known ALS genes—six people who have familial ALS caused by mutations in superoxide dismutase 1, and three people with mutations in valosin-containing protein. Sure enough, the exome-wide analysis found more variation in these two genes in ALS cases than controls. It also identified matrin 3 as an ALS gene, further validating the technique.
The analysis also identified a new ALS gene candidate in TUBA4A. No controls carried the TUBA4A variants that are predicted to disrupt the protein, but four cases did. To check their results, the authors repeated the analysis in a new dataset with 272 familial ALS cases and 5,510 controls. Again, TUBA4A stood out. The researchers found two variants in people with ALS and two in the much larger control group, indicating a far higher frequency of variation in people with the disease.
"This manuscript provides a novel approach to identifying ALS-linked genes," Aaron Gitler of Stanford University in Palo Alto, California, wrote in an email to Alzforum. "The race is now on to figure out how tubulin fits into ALS pathogenesis, and the field must now assess the TUBA4A gene in additional ALS patient cohorts." (See full comment below.)
Landers and colleagues also identified a handful of genes that were enriched in ALS cases, but did not pass statistical tests for significance. These include genes for serine/threonine kinase 24 (STK24), which participates in apoptosis, and a protein that goes by the mouthful leucine rich repeat (in flightless I homolog ) interacting protein 1 (LRRFIP1), a DNA- and RNA-binding protein. With analysis of more cases, those genes might prove significant, Landers suggested. In fact, neither VCP nor matrin 3 was statistically significant in this cohort.
Overall, the authors found seven different TUBA4A variants in people with familial ALS. Six of these were predicted to damage the protein. In one person with sporadic ALS they also discovered an eighth variant, a valine rather than a glycine at position 43. PolyPhen-2 predicted this substitution was unlikely to alter protein function. One of the six proposed dysfunctional variants also occurred in control exomes, while another showed up in one person with familial ALS but not in that person’s cousin, who also had the disease. These two might also be neutral variants, the authors suggested. Most of the variants occurred in the carboxyl end of the protein, which makes contact with other tubulin subunits and associated proteins, such as kinesins (see image above).
“If the results are independently replicated, then it would be clear that the TUBA4A gene is involved in ALS,” commented Rita Guerreiro of University College London, who was not involved in the study. “However, it is unclear what role each of the variants identified has in disease.” (See full comment below.)
TUBA4A encodes one of eight human α-tubulins, which polymerize with β-tubulins to make the microtubule cytoskeleton. TUBA4A is highly expressed in brain. Landers said the researchers immediately surmised that mutations might have a dominant-negative effect, by interfering with assembly or stability of the microtubule network. Fallini and colleagues studied microtubule dynamics of several of the variants. In mouse primary motor neurons, the wild-type and G43V TUBA4A assembled into microtubules normally. The other variants did not integrate into microtubules as efficiently, and unlike normal tubulin, they also appeared diffusely throughout the cytosol. A premature stop codon mutant, tryptophan-407-X, did not form part of microtubules, and instead made ubiquitinated cytoplasmic inclusions in many cells.
Fallini observed that some of the mutants also destabilized the microtubule cytoskeleton, and interfered with the assembly of the centrosome, the cell’s microtubule organizing center. The authors conclude that the variants of TUBA4A associated with ALS weaken the microtubule network. Landers speculated that the mutants might hamper the incorporation of normal tubulin or slow microtubule extension.
There is precedent for cytoskeletal genes being involved in neurodegeneration. At least seven other tubulins have been linked to neurological “tubulinopathies” (reviewed in Breuss and Keays, 2014). Microtubule-associated proteins also have been implicated in neurological disease. Mutations in spastin cause hereditary spastic paraplegia (Wharton et al., 2003) and tau mutations cause some cases of frontotemporal dementia. FTD and ALS share disease genes, and people with one condition often exhibit symptoms of the other malady. Notably, two of the ALS cases in the study also exhibited dementia. Guerreiro suggested it might be worth screening for TUBA4A mutations in people with FTD.
Landers and colleagues are not the only ones finding novel ways to identify ALS genes. Gitler has hunted for ALS-linked mutations without relying on large kindreds by focusing on “trios,” people with sporadic ALS and their parents (see May 2013 news story). Writing in the October 2014 PLoS Genetics, Gitler, first author Julien Couthouis, and colleagues from Stanford describe their latest strategy. Rather than looking at the whole exome, they took a candidate approach, sequencing the exons of 169 genes known or suspected to contribute to ALS. Like the authors of the Neuron study, they looked for genes that displayed more variation in 242 cases than in 129 controls. “The method is not very good for gene discovery, but for candidate screening it is perfect,” said Couthouis. "You just have to sequence what you need.”
The researchers found several novel variants. For example, they observed that several point mutations in the ataxin-2 gene occur more frequently in ALS. This supports prior analyses that have linked repeat expansions in ataxin-2 enhance to ALS risk (see Aug 2010 news story; Neuenschwander et al., 2014). They also found variants of a gene called ALS2, which has been associated with juvenile disease, in their cases. That suggests the gene could contribute to adult ALS risk, as well. The authors also identified more variants of genes they picked out in the trio study. These and other novel variants are likely, but not proven, to be ALS-associated genes, said Couthouis.—Amber Dance
References
News Citations
- The Power of Three: Genetic Trios Yield ALS Gene Candidates
- ALS—A Polyglutamine Disease? Mid-length Repeats Boost Risk
Paper Citations
- Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, Pliner HA, Abramzon Y, Marangi G, Winborn BJ, Gibbs JR, Nalls MA, Morgan S, Shoai M, Hardy J, Pittman A, Orrell RW, Malaspina A, Sidle KC, Fratta P, Harms MB, Baloh RH, Pestronk A, Weihl CC, Rogaeva E, Zinman L, Drory VE, Borghero G, Mora G, Calvo A, Rothstein JD, ITALSGEN Consortium, Drepper C, Sendtner M, Singleton AB, Taylor JP, Cookson MR, Restagno G, Sabatelli M, Bowser R, Chiò A, Traynor BJ. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014 May;17(5):664-6. Epub 2014 Mar 30 PubMed.
- Panoutsopoulou K, Tachmazidou I, Zeggini E. In search of low-frequency and rare variants affecting complex traits. Hum Mol Genet. 2013 Oct 15;22(R1):R16-21. Epub 2013 Aug 6 PubMed.
- Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol. 2011 Sep 14;12(9):227. PubMed.
- Breuss M, Keays DA. Microtubules and neurodevelopmental disease: the movers and the makers. Adv Exp Med Biol. 2014;800:75-96. PubMed.
- Wharton SB, McDermott CJ, Grierson AJ, Wood JD, Gelsthorpe C, Ince PG, Shaw PJ. The cellular and molecular pathology of the motor system in hereditary spastic paraparesis due to mutation of the spastin gene. J Neuropathol Exp Neurol. 2003 Nov;62(11):1166-77. PubMed.
- Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM. Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol. 2014 Dec;71(12):1529-34. PubMed.
External Citations
Further Reading
Papers
- Maximino JR, de Oliveira GP, Alves CJ, Chadi G. Deregulated expression of cytoskeleton related genes in the spinal cord and sciatic nerve of presymptomatic SOD1(G93A) Amyotrophic Lateral Sclerosis mouse model. Front Cell Neurosci. 2014;8:148. Epub 2014 May 26 PubMed.
- Julien JP, Millecamps S, Kriz J. Cytoskeletal defects in amyotrophic lateral sclerosis (motor neuron disease). Novartis Found Symp. 2005;264:183-92; discussion 192-6, 227-30. PubMed.
- Cairns NJ, Lee VM, Trojanowski JQ. The cytoskeleton in neurodegenerative diseases. J Pathol. 2004 Nov;204(4):438-49. PubMed.
- Farah CA, Nguyen MD, Julien JP, Leclerc N. Altered levels and distribution of microtubule-associated proteins before disease onset in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2003 Jan;84(1):77-86. PubMed.
News
- Evidence Mounts That Mitochondrial Gene Is Bona Fide ALS, FTD Risk Factor
- TREM2 Variant Doubles the Risk of ALS
- Researchers Revel in C9ORF72 Advances at RNA Symposium
- Study Links Motor Neuron Disease to New Receptor Tyrosine Kinase
- One, Two, Three…Four? Peripherin Jams With Neurofilament in Quartet
- Profilin Gene Is Actin’ in ALS
Primary Papers
- Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, Scotter EL, Kost J, Keagle P, Miller JW, Calini D, Vance C, Danielson EW, Troakes C, Tiloca C, Al-Sarraj S, Lewis EA, King A, Colombrita C, Pensato V, Castellotti B, de Belleroche J, Baas F, ten Asbroek AL, Sapp PC, McKenna-Yasek D, McLaughlin RL, Polak M, Asress S, Esteban-Pérez J, Muñoz-Blanco JL, Simpson M, SLAGEN Consortium, van Rheenen W, Diekstra FP, Lauria G, Duga S, Corti S, Cereda C, Corrado L, Sorarù G, Morrison KE, Williams KL, Nicholson GA, Blair IP, Dion PA, Leblond CS, Rouleau GA, Hardiman O, Veldink JH, van den Berg LH, Al-Chalabi A, Pall H, Shaw PJ, Turner MR, Talbot K, Taroni F, García-Redondo A, Wu Z, Glass JD, Gellera C, Ratti A, Brown RH Jr, Silani V, Shaw CE, Landers JE, SLAGEN Consortium. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014 Oct 22;84(2):324-31. PubMed.
- Couthouis J, Raphael AR, Daneshjou R, Gitler AD. Targeted exon capture and sequencing in sporadic amyotrophic lateral sclerosis. PLoS Genet. 2014 Oct;10(10):e1004704. Epub 2014 Oct 9 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Stanford University
The last 10 years have seen several major breakthroughs in ALS genetics, many resulting from genetic linkage studies in large, multigenerational families with the disease. However, these families are very rare and few remain that haven’t yet been analyzed for linkage with known ALS genes. Here, Landers and colleagues take a novel approach to circumvent this problem, performing exome sequencing on 363 familial ALS (fALS) cases and 31 related controls, and then subjecting the sequencing data to a genome-wide association study to identify genes with an enrichment of rare, potentially damaging variants in fALS cases compared to controls. They spiked in samples from patients who they knew had SOD1 mutations as a control and pulled these out as the top hit from the screen.
The next-highest-ranking gene they identified is TUBA4A, encoding the Tubulin, alpha 4A protein. The identified association in this gene was due to five patients in their fALS cohort, each with a different non-synonymous mutation in highly-conserved residues of the gene. Critically, they go on to confirm this association by analyzing an independent set of 272 FALS cases and many thousands of controls.
This paper, especially now that there are several large-scale international efforts at sporadic ALS exome sequencing underway, provides an excellent example of how to analyze the data to discover important new genetic associations, and to validate the discoveries with functional data. Beyond that, it seems that the tubulin mutations Landers and colleagues have discovered are going to be important for ALS in general. In my opinion, this manuscript provides a novel approach to identifying ALS-linked genes and may have identified one new ALS-associated gene, TUBA4A. The race is now on to figure out how tubulin fits into ALS pathogenesis and the field must now assess the TUBA4A gene in additional ALS patient cohorts.
View all comments by Aaron GitlerVan Andel Institute
Smith et al. performed exome sequencing in a cohort of 363 index cases from ALS families (FALS). They used the control exome data from the NHLBI’s Exome Variant Server (n=4,300 European Americans) to compare aggregated counts of minor alleles in gene windows between cases and controls. Results showed a significant excess of TUBA4A in patients. This difference between cases and controls was replicated by studying an additional cohort of 272 FALS and 5,510 controls. Subsequent functional analyses revealed the destabilization of microtubules by the TUBA4A mutations identified.
This is an interesting study making good use of publicly available exome sequencing-derived data. Although the cohorts of patients studied were small, the spike in a number of cases with known mutations (particularly, SOD1) and the use of these cases as positive controls for the genetic methods give robustness to the results.
As the authors recognize, the main drawback of the aggregation of variants for comparison between cases and controls is the difficulty of attributing pathogenicity to specific variants. If the current results get to be independently replicated, it is clear that the TUBA4A gene is involved in ALS, however, it is rather unclear what role each of the variants identified has in disease. The authors refer to a set of eight variants (p.G43V, p.T145P, p.R215C, p.R320C/H, p.A383T, p.W407X and p.K430N); two of these (p.G43V and p.A383T) were found in controls in the replication stage.
Looking at the data recently released by the Exome Aggregation Consortium (ExAC), which includes data derived from exome sequencing of more than 63,000 samples, a few of the mutations reported as possibly involved in ALS can be found in one or two individuals (p.G43V, one allele; p.R320C, two allele; and p.A383T, one allele). In particular, it is difficult to reconcile the genetic and functional data for the variant p.A383T that the authors detected in a control sample (as they did for p.G43V), but functionally shows significant impacts on microtubule polymerization and microtubule network stabilization.
As the authors discuss, it is interesting to note that mutations in other members of the tubulin family cause both neurodevelopmental and neurodegenerative disorders, with defects responsible for the former occurring in proteins that are highly expressed during brain development but then wane with age, and the latter being associated with genes increasingly expressed with age. It is also interesting that two of the cases with TUBA4A mutations have frontal cognitive decline in addition to ALS, spotlighting the potential for gene screens in FTD cohorts to turn up TUBA4A as well.
View all comments by Rita GuerreiroMake a Comment
To make a comment you must login or register.