Nilvadipine Fails to Slow Cognitive Decline in AD Patients
Quick Links
A putative amyloid-lowering agent, the blood pressure drug nilvadipine has proven ineffective in people with mild to moderate Alzheimer’s disease in a Phase 3 trial. In the September 24 PLoS Medicine, researchers led by Brian Lawlor at St. James’ Hospital, Dublin, reported that 18 months of treatment did not slow cognitive decline on the primary endpoints, the ADAS-Cog12 and the CDR sum of boxes. Biomarker analyses have not yet been published, so it is unclear if the drug hit its biologic targets.
- Nilvadipine did not slow cognitive decline in mild to moderate AD.
- Researchers are still analyzing biomarkers of target engagement.
Although nilvadipine lowers blood pressure by blocking calcium channels, this is thought to be unrelated to amyloid plaque reduction. In cell and animal models, nilvadipine suppresses Aβ production and boosts clearance of the peptide across the blood-brain barrier (Jul 2011 news). Mouse studies also indicate that nilvadipine inhibits the spleen tyrosine kinase (SYK), which activates the tau kinase GSK-3β, hence the drug potentially reduces tau phosphorylation, as well. In that same study, researchers reported that a known SYK inhibitor mimicked the effects of nilvadipine on amyloid and tau (Paris et al., 2014). Whether the purported amyloid and tau effects are related is unknown.
In the Phase 3 trial, which began in 2013, researchers used a dose of nilvadipine, 8 mg once daily, that has minimal effects on blood pressure. Researchers recruited 511 participants from nine European countries, with 253 of them randomized to take nilvadipine, the rest placebo. Treatment and placebo groups were well-matched overall, although the treatment group had about twice as many diabetics. Participants had an average age of 73 and MMSE of 20 and met NINCDS-ADRDA criteria for Alzheimer’s, but the diagnosis was not confirmed with biomarkers.
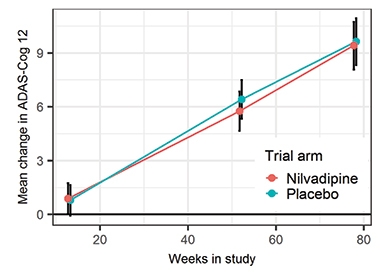
Nil Effect.
In people on nilvadipine or placebo, cognition worsened at the same rate. [Courtesy of PLoS Medicine, Lawlor et al.]
No safety issues emerged. There were slightly more adverse events in the treatment group, but these were judged unrelated to the drug. About 10 percent of participants dropped out in both the treatment and placebo groups. The treatment only modestly lowered blood pressure, with median systolic pressure falling by five points.
The nilvadipine trial was powered to detect a 50 percent slowing of cognitive decline on the primary endpoints, but found no difference (see image). In addition, the researchers saw no change in the Disability Assessment for Dementia, which measures functional outcomes. In prespecified subgroup analyses, treated participants with an MMSE above 20 appeared to decline more slowly than the placebo group, but those with a lower MMSE declined faster on drug. Males and ApoE4 carriers also declined more slowly on drug than on placebo. However, the numbers in these subgroups were too small to be statistically meaningful. Biomarker analyses of Aβ levels in cerebrospinal fluid and blood, as well as MRI data on cerebral blood flow, are ongoing. The researchers did not measure effects on SYK, so it is unclear if 8 mg was enough to inhibit the kinase.
Lawlor suggested that because nilvadipine lowers amyloid, it might work better at preclinical disease stages, a point echoed by Costantino Iadecola at Weill Cornell Medical College in New York. “It is becoming eminently clear that in AD, as in other brain disease, acute or chronic, the earlier the intervention the better,” he wrote to Alzforum. He was not involved in the study. “I hope that funding can be obtained to repeat the study in presymptomatic biomarker-positive patients, an intervention that is likely to have a greater impact,” Iadecola added.
The nilvadipine formulation used contains two mirror-image enantiomers of the same drug. Both inhibit SYK, but only the plus enantiomer blocks calcium channels and lowers blood pressure. Lawlor is considering testing only the minus enantiomer in patients at an earlier disease stage. Because this form has no effect on blood pressure, researchers could use a higher dose.—Madolyn Bowman Rogers
References
News Citations
Paper Citations
- Paris D, Ait-Ghezala G, Bachmeier C, Laco G, Beaulieu-Abdelahad D, Lin Y, Jin C, Crawford F, Mullan M. The spleen tyrosine kinase (Syk) regulates Alzheimer amyloid-β production and Tau hyperphosphorylation. J Biol Chem. 2014 Dec 5;289(49):33927-44. Epub 2014 Oct 20 PubMed.
External Citations
Further Reading
Primary Papers
- Lawlor B, Segurado R, Kennelly S, Olde Rikkert MG, Howard R, Pasquier F, Börjesson-Hanson A, Tsolaki M, Lucca U, Molloy DW, Coen R, Riepe MW, Kálmán J, Kenny RA, Cregg F, O'Dwyer S, Walsh C, Adams J, Banzi R, Breuilh L, Daly L, Hendrix S, Aisen P, Gaynor S, Sheikhi A, Taekema DG, Verhey FR, Nemni R, Nobili F, Franceschi M, Frisoni G, Zanetti O, Konsta A, Anastasios O, Nenopoulou S, Tsolaki-Tagaraki F, Pakaski M, Dereeper O, de la Sayette V, Sénéchal O, Lavenu I, Devendeville A, Calais G, Crawford F, Mullan M, NILVAD Study Group. Nilvadipine in mild to moderate Alzheimer disease: A randomised controlled trial. PLoS Med. 2018 Sep;15(9):e1002660. Epub 2018 Sep 24 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Weill College Medicine
There was a strong rationale for this trial, considering the strength of the preclinical studies suggesting improvement of amyloid pathology in mouse models, and the evidence from observational studies in humans, leaning toward cognitive improvement. The double-blinded placebo-controlled design of the trial is also a strength.
The negative outcome suggests several scenarios:
1. Inhibition of dihydropyridine-sensitive Ca2+ channels is not effective in AD;
Or:
2. The dose of the drug was too low;
3. The treatment was initiated too late in the course of the disease;
4. AD diagnosis was not confirmed by biomarkers, resulting in the inclusion of a large number of patients without AD pathology (plaques and tangles).
The last two scenarios are likely to have had the greatest impact on the outcome. The drug, in theory, could have been effective by clearing amyloid or by preventing the deleterious effects of hypertension on AD pathology, but these factors start to affect the brain in midlife prior to the patients becoming symptomatic. Therefore, in a sense, it is not surprising that no effect on cognition was observed. On the positive side, a hint of effectiveness was observed in the less-severely affected male subgroup, highlighting a potential window of opportunity earlier in the disease process. I hope that funding can be obtained to repeat the study in presymptomatic biomarker-positive patients in the early phases of the disease process. Such an intervention is likely to have a greater impact on the subsequent cognitive decline.
It is becoming eminently clear that in AD, as in other brain disease, acute or chronic, the earlier the intervention the better. The challenge is going to be to identify the patients at the “right" stage of disease, amenable to intervention.
Mayo Clinic College of Medicine
This fully powered, 18-month study of nilvadipine (NILVAD) by Brian Lawlor and colleagues in persons with clinically diagnosed AD dementia failed to show any cognitive or functional benefits of NILVAD. The trial was well-designed and the published report is well-written. Unfortunately, once again, a promising idea has failed to translate into therapeutic benefit.
The rationale for NILVAD originated from several observations: 1) that calcium channel blockers have been implicated in dementia prevention in large-scale, anti-hypertensive drug trials, 2) that NILVAD itself might have some actions on Alzheimer amyloid and tau production or clearance.
In thinking about why the trial failed to show benefits, I wonder whether its inclusion criteria appropriately matched the proposed mechanisms of action (MOA) attributed to NILVAD. That is, if the investigators thought that the MOA of NILVAD was cerebrovascular, shouldn’t they have required all participants to have some degree of overt cerebrovascular disease? Or, if they thought that the mechanisms were Alzheimer-related, shouldn’t they have required some biomarker confirmation? As it is, their study group were neither overrepresented with a high burden of cerebrovascular disease, nor purely an AD group (i.e. plaques and tangles etiology).
This brings up a question that perhaps has been too long left unaddressed in dementia therapeutics: How important is it that the target of the MOA be present in the study population? It seems obvious that the answer ought to be “yes,” but restricting the study population is costly and difficult. As the field continues to be confronted by failure, is there any choice? To be sure, having the right patients is only one of the necessary ingredients: Having the right MOA, the right dose, and the right timing are essential. All that said, I must congratulate the investigators on the rigor of their implementation and execution of this trial.
KULeuven
The AD therapeutic problem has become a diagnostic one: How to define and detect prodromal AD? Even more important: How to stratify the different types of AD early in the disease process? A daunting task we must recognize and attack.
Liverpool John Moores University
Several points have been made as to why this drug may have failed; however, here is one comment I have not seen made.
Outcome measures such as the ADAS-COG, MMSE and ADCS-ADL, which are typically used in these trials, are broad measures of overall cognitive ability and behavioral function. They are excellent when attempting to provide a broad picture of how a person is performing at a certain point in time, but they encompass a multitude of cognitive processes. Some of the processes included in these measures are highly affected by pathology, but others not so much. These latter processes/functions are less likely to change as a consequence of increased or decreased pathology, and they will do so at different speeds. Therefore, while these measures may be able to pick up some changes due to the intervention, these changes may be small and largely drown out into a larger mass of data that are not sensitive.
Why are finer cognitive measures not applied to these trials more frequently and systematically, and/or extracted from existing data? I am, of course, not alone in asking this, and various solutions have been put forward, although they typically seem to again involve measures of performance that combine multiple sources of cognitive inputs (e.g., Wessels et al., 2015; Wessels et al., 2018).
I am thinking of recall measured based on the analysis of serial position, which has been found to be sensitive to subtle changes in cognitive ability in the elderly. For instance, examining delayed primacy or the recency ratio (e.g., Bruno et al., 2013; Bruno et al., 2018), both of which can be extracted from the ADAS-cog with some work (and some caveats); also measures of associative memory. More simply, why not interrogate the memory data in a test like the ADAS-cog more specifically, and delayed recall data in particular?
I realize none of this would have been planned in advance, and as post-hoc analyses suffer from potential type 1 error rate inflation, but still I feel this is worth pursuing when assessing the effectiveness of these interventions. I am not suggesting that, in the first instance, different tests are used to provide outcome measures, but that existing data are looked at in different ways.
There is a conceptual spectrum, ranging from functional and structural neuroimaging markers on one end, which may be sensitive to treatment but may not adequately represent actual changes in function and quality of life, to ADAS cog on the other end, which represents global functioning but may not be sensitive to subtle change. In the middle, there are other options, which may be an ideal compromise, but these seem to be generally neglected.
References:
Bruno D, Koscik RL, Woodard JL, Pomara N, Johnson SC. The recency ratio as predictor of early MCI. Int Psychogeriatr. 2018 Apr 18;:1-6. PubMed.
Bruno D, Reiss PT, Petkova E, Sidtis JJ, Pomara N. Decreased recall of primacy words predicts cognitive decline. Arch Clin Neuropsychol. 2013 Mar;28(2):95-103. PubMed.
Wessels AM, Andersen SW, Dowsett SA, Siemers ER. The Integrated Alzheimer's Disease Rating Scale (iADRS) Findings from the EXPEDITION3 Trial. J Prev Alzheimers Dis. 2018;5(2):134-136. PubMed.
Wessels AM, Siemers ER, Yu P, Andersen SW, Holdridge KC, Sims JR, Sundell K, Stern Y, Rentz DM, Dubois B, Jones RW, Cummings J, Aisen PS. A Combined Measure of Cognition and Function for Clinical Trials: The Integrated Alzheimer's Disease Rating Scale (iADRS). J Prev Alzheimers Dis. 2015 Dec 1;2(4):227-241. PubMed.
Previous commentators have explained the failure of nilvadipine by attributing it to infelicitous patient selection. I believe it failed because what it targeted is not the actual “cause” of AD.
Can we now begin to seriously explore other triggers, which will open different therapeutic pathways? There are several of these ripe for attention, including infectious agents, my personal bias.
Make a Comment
To make a comment you must login or register.