New Target for Tauopathies? Blocking Nuak1 Could Reduce Tau Build-Up
Quick Links
In the Alzheimer’s brain, tau dissociates from microtubules, becomes hyperphosphorylated, and aggregates into neurofibrillary tangles. What kicks off this process? In the October 6 Neuron, researchers led by Huda Zoghbi and Juan Botas at the Jan and Dan Duncan Neurological Research Institute at Texas Children’s Hospital, Houston, indict a new suspect. The kinase Nuak1, which structurally resembles AMP-activated protein kinase (AMPK), phosphorylates serine 356 in tau’s microtubule-binding domain, and this modification prevents tau degradation, causing levels to build up in the cytoplasm. That in turn triggers hyperphosphorylation, the authors report. Cutting Nuak1 levels in half in a tauopathy mouse model ameliorated pathology, restored normal synaptic plasticity, and improved memory. The findings point toward a new strategy for tackling tau accumulation, Zoghbi suggested.
Others agreed. “Therapeutic targeting of AMPK-related kinases … may offer a new way to combat AD and related tauopathies, for which there are currently no effective treatment options,” Bingwei Lu at Stanford University, Palo Alto, California, wrote to Alzforum. Franck Polleux at Columbia University, New York City, called the paper exciting. “The authors present strong evidence that Nuak1 is an important tau kinase, and may be a promising therapeutic target,” he said.
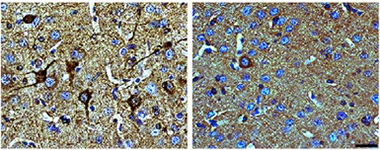
Fewer Tangles.
Tau transgenic mice with half the normal level of Nuak1 (right) accumulate far fewer neurofibrillary tangles (brown) in cortex than do control transgenics (left). Nuclei are blue. [Courtesy of Neuron, Lasagna-Reeves et al., 2016.]
Zoghbi’s earlier work on spinocerebellar ataxias convinced her that protein levels matter in neurodegeneration, she told Alzforum. Previously, she had reported that lowering ataxin1 protein in a mouse model of the disease protected neurons and improved motor skills (see Park et al., 2013). Zoghbi wondered if the same approach might help in tauopathies such as Alzheimer’s disease, since previous work associated less tau with better memory in AD mice (see May 2007 news).
To find factors that influence the amount of tau, Zoghbi’s group focused on kinases. They screened all known human kinases by knocking them out one by one with RNAi in a human cell line, and measuring the resulting tau levels. Meanwhile, Botas’ group ran a similar screen in fruit flies that overexpress human tau, looking for kinases that lessened degeneration in the eye. The authors identified 16 kinases that affected both systems. From these, they selected Nuak1 for follow-up based on its particularly robust effects. Nuak1 was not previously known to interact with tau. Past screens for tau modifiers have mostly focused on hyperphosphorylation or aggregation of the protein, rather than abundance, which may be why this kinase did not show up in those studies, the authors noted.
First author Cristian Lasagna-Reeves turned to in vitro assays to validate Nuak’s interaction with tau, finding that it selectively acted on only Ser356. This site and its neighbor Ser262 have been dubbed “priming sites,” because their phosphorylation leads tau to detach from microtubules and subsequently become hyperphosphorylated. In keeping with this result, Lasagna-Reeves and colleagues found that overexpression of Nuak1 in neuroblastoma cells pumped up Ser356 phosphorylation as well as levels of tau and hyperphosphorylated tau, while depleting Nuak1 did the opposite. The increase in overall phosphorylation supports the idea that phosphorylation at Ser356 kicks off a cascade, the authors note.
Why did tau levels soar? The authors saw no difference in tau mRNA levels, indicating no change in gene expression. Instead, tau phosphorylated at Ser356 hung around longer. Using the cell assays, the authors found that phosphorylation at this site prevented the ubiquitinase CHIP from binding and tagging the protein for degradation, as it normally does (see Dickey et al., 2007; Dickey et al., 2008). Meanwhile, cells that expressed a mutant version of tau lacking the Ser356 site were unaffected by overexpression of Nuak1.
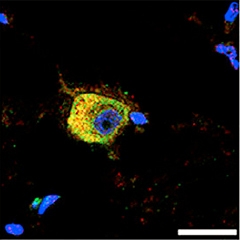
Close Association.
Nuak1 (red) lingers around neurofibrillary tangles (green; overlay appears yellow) in neurons from a brain with progressive supranuclear palsy. [Courtesy of Neuron, Lasagna-Reeves et al., 2016.]
To probe Nuak1 in a disease model, the authors crossed Nuak1 heterozygous knockouts with P301S mice, which express human mutant tau at five times endogenous levels. These animals develop neurofibrillary tangles by six months of age. However, the Nuak1-deficient mice sported less pathological tau than their P301S littermates at 7.5 months, having about one-third as many neurons that contained paired helical filaments (see image above). In addition, their total tau levels were down about 20 percent, and they had about half as much tau phosphorylated at Ser356.
The changes in tau came with altered behavior, too. At 7.5 months old, Nuak1-deficient mice remembered where the hidden platform was in the Morris water maze better than their P301S littermates did, although less well than controls. Likewise, they froze more than PS01Ss in an environment where they had previously received a shock, though again not to control levels. Both these tasks depend on the hippocampus. The authors tested hippocampal function directly by implanting electrodes and measuring synaptic plasticity in live, active mice. While P301S animals were impaired, their Nuak1-deficient littermates were indistinguishable from controls.
Does Nuak1 play a role in human disease? In postmortem brain tissue, the authors detected twice as much Nuak1 protein in AD brains as in healthy controls. They also measured double the normal Nuak1 in brains from people who had had progressive supranuclear palsy, a different tauopathy, suggesting elevated Nuak1 may be common to tauopathies. S356 phosphorylation by Nuak1 may be broadly activated in different neurodegenerative diseases, they posit. In addition, Nuak1 localized to neurofibrillary tangles in both conditions (see image above).
Polleux found the human data intriguing, noting that the findings mirror those for AMPK. He had previously reported that AMPK phosphorylates tau at Ser262, and that this kinase associates with tau tangles in AD brains. AD brains contain an abundance of the activated form of AMPK, Polleux and colleagues reported. In mice, they uncovered evidence that Aβ oligomers switch on AMPK (see Apr 2013 news). It would be interesting to know if Aβ also overactivates Nuak1, or other members of the same kinase family, Polleux said. Lasagna-Reeves plans to study Nuak1 and Aβ interactions by crossing the heterozygous Nuak1 knockouts with an APP mouse model.
Meanwhile, Nuak1 might make an attractive therapeutic target, Zoghbi agreed. There are no drugs that inhibit Nuak1, and researchers would have to be careful not to suppress it too much to spare potentially crucial functions. For example, Nuak1 controls terminal axon branching during development (see Courchet et al., 2013). More broadly, Zoghbi believes the general strategy of lowering tau may be useful. She is following up on other kinases uncovered in this screen, and plans to screen a much broader group of proteins for their effects on tau as well.—Madolyn Bowman Rogers
References
News Citations
- APP Mice: Losing Tau Solves Their Memory Problems
- Tracing a Path from Aβ to Tau Leads Scientists to Lost Synapses
Research Models Citations
Paper Citations
- Park J, Al-Ramahi I, Tan Q, Mollema N, Diaz-Garcia JR, Gallego-Flores T, Lu HC, Lagalwar S, Duvick L, Kang H, Lee Y, Jafar-Nejad P, Sayegh LS, Richman R, Liu X, Gao Y, Shaw CA, Arthur JS, Orr HT, Westbrook TF, Botas J, Zoghbi HY. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature. 2013 Jun 20;498(7454):325-31. PubMed.
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, Ash P, Shoraka S, Zlatkovic J, Eckman CB, Patterson C, Dickson DW, Nahman NS, Hutton M, Burrows F, Petrucelli L. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J Clin Invest. 2007 Mar;117(3):648-58. PubMed.
- Dickey CA, Koren J, Zhang YJ, Xu YF, Jinwal UK, Birnbaum MJ, Monks B, Sun M, Cheng JQ, Patterson C, Bailey RM, Dunmore J, Soresh S, Leon C, Morgan D, Petrucelli L. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc Natl Acad Sci U S A. 2008 Mar 4;105(9):3622-7. PubMed.
- Courchet J, Lewis TL Jr, Lee S, Courchet V, Liou DY, Aizawa S, Polleux F. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 2013 Jun 20;153(7):1510-25. PubMed.
Further Reading
News
- Stress Granule Protein Entwines and Misfolds Tau
- ROCK’n the Tau Pathway?
- Protecting Proteasomes from Toxic Tau Keeps Mice Sharp
- Two-in-One: Single Protein Untangles and Chops Up Tau Fibrils
- New Type of Toxic Tau? Acetylated Form Correlates With Memory Defects
- Inventory of Tau Modifications Hints at Undiscovered Functions
- Chaperone “Saves” Tau, Turning it into Toxic Oligomers
- Therapeutic Approaches Target Deubiquitinase, Protein Turnover
Primary Papers
- Lasagna-Reeves CA, de Haro M, Hao S, Park J, Rousseaux MW, Al-Ramahi I, Jafar-Nejad P, Vilanova-Velez L, See L, De Maio A, Nitschke L, Wu Z, Troncoso JC, Westbrook TF, Tang J, Botas J, Zoghbi HY. Reduction of Nuak1 Decreases Tau and Reverses Phenotypes in a Tauopathy Mouse Model. Neuron. 2016 Oct 19;92(2):407-418. Epub 2016 Oct 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Max Planck Institute for Biophysical Chemistry
Cristian Lasagna-Reeves and colleagues in the lab of Huda Zoghbi show—using a variety of model systems for tauopathy—that the serine/threonine-protein kinase Nuak1 plays an important role in neurotoxicity associated with the protein tau. In addition, they demonstrate that Nuak1 associated with pathological accumulation of tau in Alzheimer’s disease and progressive supranuclear palsy (PSP). In line with the importance of Nuak1 in tau-related neurodegeneration, decreasing Nuak1 levels or activity reversed several deficits in a tauopathy mouse model. Using a variety of approaches, Lasagna-Reeves further shows that Nuak1 phosphorylates tau at S356, but not at S262, with phosphorylation of S356 playing a critical role for the proposed role of Nuak1. S356 is located in the C-terminal pseudo-repeat region of tau, which is both important for pathogenic aggregation and also binds to microtubules (MT). Identification of a kinase, which selectively phosphorylates S356, might therefore open new possibilities to influence both the physiological and pathological function of tau.
From a mechanistic point of view, the study raises an intriguing question about the role of S356 as a phosphorylation site when compared to other tau phosphorylation sites such as S262. For example, we (Schwalbe et al., 2013) have shown that phosphorylation of tau at S356 does not completely abolish binding to microtubules, but diminishes the MT-binding contribution of pseudo-repeat R4 and thus can contribute to an overall decrease in MT-affinity. At the same time, much less is known about the contribution of R4 and in particular phosphorylation at S356 for MT assembly. It will also be interesting to address in the future how Nuak1 achieves selectivity towards S356 when compared to S262, because both residues are part of a KIGS-motif, which can be phosphorylated 100 percent by MARK2 (Schwalbe et al., 2013). The selective nature of Nuak1 phosphorylation at S356 also raises the question about the potential contribution of individual pseudo-repeats and individual phosphorylation sites within the four repeats of tau for tau-mediated neurotoxicity.
References:
Schwalbe M, Biernat J, Bibow S, Ozenne V, Jensen MR, Kadavath H, Blackledge M, Mandelkow E, Zweckstetter M. Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry. 2013 Dec 17;52(50):9068-79. Epub 2013 Nov 22 PubMed.
Stanford University
In this interesting study by Dr. Huda Zoghbi and colleagues, the authors showed that the AMPK-related kinase Nuak1 regulates tau toxicity through controlling its stability in mammalian cells and in Drosophila and mouse tauopathy models. The authors further showed that Nuak1 does so by phosphorylating tau at the S356 site in the fourth microtubule-binding domain (MBD). Functionally, 50 percent reduction of Nuak1 activity effectively reduced tau phosphorylation and stability and the neuropathological and behavioral phenotypes in the P301S tauopathy mouse model. The clinical relevance of the study is highlighted by the analysis of Alzheimer’s disease (AD) and progressive surpranuclear palsy patient brain samples, where Nuak1 is found associated with insoluble tau in neurofibrillary tangles (NFT).
This study, together with previous studies on phosphorylation of the MBD by PAR-1, another AMPK-related kinase, highlights the importance of tau phosphorylation at S262 and S365 in conferring tau toxicity (Nishimura et al., 2004), stability (Ando et al., 2016; Ando et al., 2016), and in mediating the toxic effect of Aβ42 (Yu et al., 2012; Ando et al., 2016), and emphasizes the importance of AMPK-related kinase in tau pathogenesis. The phosphorylation of the MBD by these kinases early in the disease process may lead to tau dissociation from the microtubule, its stabilization, hyperphosphorylation by downstream kinases, and the formation of NFTs. Therapeutic targeting of these AMPK-related kinases, or their upstream activating kinase LKB1, which has been shown to regulate tau toxicity through PAR-1 in Drosophila (Wang et al., 2007), may offer a new way to combat AD and related tauopathies, for which there are currently no effective treatment option.
References:
Nishimura I, Yang Y, Lu B. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell. 2004 Mar 5;116(5):671-82. PubMed.
Ando K, Oka M, Ohtake Y, Hayashishita M, Shimizu S, Hisanaga S, Iijima KM. Tau phosphorylation at Alzheimer's disease-related Ser356 contributes to tau stabilization when PAR-1/MARK activity is elevated. Biochem Biophys Res Commun. 2016 Sep 16;478(2):929-34. Epub 2016 Aug 9 PubMed.
Ando K, Maruko-Otake A, Ohtake Y, Hayashishita M, Sekiya M, Iijima KM. Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Aβ42-Induced Tau Toxicity. PLoS Genet. 2016 Mar;12(3):e1005917. Epub 2016 Mar 29 PubMed.
Yu W, Polepalli J, Wagh D, Rajadas J, Malenka R, Lu B. A critical role for the PAR-1/MARK-tau axis in mediating the toxic effects of Aβ on synapses and dendritic spines. Hum Mol Genet. 2012 Mar 15;21(6):1384-90. PubMed.
Ando K, Maruko-Otake A, Ohtake Y, Hayashishita M, Sekiya M, Iijima KM. Stabilization of Microtubule-Unbound Tau via Tau Phosphorylation at Ser262/356 by Par-1/MARK Contributes to Augmentation of AD-Related Phosphorylation and Aβ42-Induced Tau Toxicity. PLoS Genet. 2016 Mar;12(3):e1005917. Epub 2016 Mar 29 PubMed.
Wang JW, Imai Y, Lu B. Activation of PAR-1 kinase and stimulation of tau phosphorylation by diverse signals require the tumor suppressor protein LKB1. J Neurosci. 2007 Jan 17;27(3):574-81. PubMed.
Make a Comment
To make a comment you must login or register.