Mutant Tau Stiffens Axon Cytoskeleton Near Soma
Quick Links
Changes to the microtubule-binding protein tau cause it to fall off these struts and aggregate, forming neurofibrillary tangles. But is that the reason for tau toxicity? A study in the September 18 Neuron suggests that V337M tau, a variant associated with frontotemporal dementia, causes morphological changes at the base of the axon. Scientists led by Li Gan, then at the Gladstone Institute of Neurological Disease, San Francisco, found that neurons derived from patients with the mutation are hyperexcitable. They had unusually short, unresponsive axon initial segments (AIS), which usually initiate action potentials and steer neuronal plasticity. Gan’s results suggest that this FTD mutation robs neurons of a mechanism for maintaining electrical homeostasis.
- The V337M mutation tightens tau’s grip on the cytoskeletal protein EB3.
- This locks down the axon initial segment, affecting plasticity.
- Neurons become hyperexcitable.
“This was a nicely executed study that showed pathological tau can influence the axon initial segment,” said Joanna Jankowsky, Baylor College of Medicine, Houston. “There are parts of the neuron we hadn’t been thinking about that may be influenced by tau.”
“The initiation of axon potential firing has received little attention in neurodegeneration research,” said Jürgen Götz, “What I really like about this paper is the use of complementary techniques to mechanistically dissect the effect of FTD mutant tau on neuronal function.”
Because many FTD-causing tau mutations occur in the microtubule-binding domain, scientists believed they lead to disease by weakening microtubules (Rossi and Tagliavini, 2015; Hong et al., 1998). However, studies have mostly found that removing tau leaves microtubules intact (Roberson et al., 2007). On the other hand, FTD patients have hyperexcitable neurons, seizures, and highly synchronized neuronal networks (Beagle et al., 2017). Could the mutant protein alter neuronal excitability in some way?
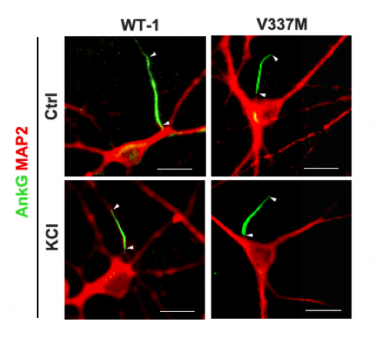
Plasticity Deficit. In iPSC-derived wild-type neurons (left) the axon initial segment (green) is long initially (top)and shrinks after chronic depolarization with KCl (bottom). In neurons with V337M tau (right), the AIS starts out and remains short. [Courtesy of Sohn et al., 2019.]
To find out, first author Peter Dongmin Sohn and colleagues focused on the AIS. The closest part of the axon to the soma, the AIS contains a high concentration of voltage-gated ion channels and triggers action potentials. It regulates neuronal excitability by lengthening or shrinking in response to less or more activity, respectively. This remodeling relies on a reorganization of the cytoskeleton, in particular ankyrin G. Staining for AnkG revealed that the AIS shrank, making it less excitable, when Sohn and colleagues depolarized wild-type neurons for two days (see image at left). In contrast, in iPSC-derived neurons from a patient with the V337M tau mutation, the AIS was about 20 percent shorter to begin with, and chronic depolarization did not change its length. This suggested the region was less plastic. If the researchers used CRISPR-Cas9 to correct the mutation, then the initial length of the AIS and its plasticity matched that of wild-type cells.
The researchers next compared electrophysiological properties of mutant and control neurons. In culture, neurons carrying mutant tau more often fired in synch, having longer network bursts containing more spikes, than did wild-type neurons. This suggested the mutated tau caused a type of hyper-synchrony. After a two-day depolarization, these neurons fired six times faster, while the rate stayed steady in isogenic controls.
To find out how the mutant tau might be interfering with homeostatic control of neuronal excitability, the authors examined tau binding partners in the AIS. Tau interacts with end-binding protein 3 (EB3), another component of the AIS cytoskeleton (Sayas et al., 2015). EB3 stabilizes the AIS by linking microtubules to AnkG.

Getting a Grip. At left, wild-type tau (blue) binds EB3 (green), which anchors AnkG (yellow) to microtubules (white). To the right, mutant tau (red) binds more tightly to EB3 and clamps it in place, making the AIS rigid and unchanging. [Courtesy of Sohn et al., 2019.]
Sohn and colleagues determined that tau binds EB3 directly, and that V337M tau does so more tightly. In tau V337M neurons, EB3 levels in the AIS were 40 percent higher than in wild-type. What’s more, rather than distributing throughout the AIS cytosol, EB3 gathered just under the plasma membrane, corralling AnkG. In all, the data suggested that through its grip on EB3, tau concentrated the protein in the AIS and immobilized AnkG such that it couldn’t respond to electrical activity.
Would removing EB3 rescue plasticity in V337M neurons? Suppressing translation with siRNA, Sohn reduced levels by 80 percent, which restored both the length and plasticity of the AIS. Reducing mutant tau in these neurons by 40 percent had similar effects—EB3 levels shrank in the AIS, and plasticity was restored.
“Our study provides a completely different explanation for why tau is toxic—not because it forms aggregates, but because it binds to cytoskeletal proteins important for plasticity and makes rigid structures,” Gan, who has since moved to Weill Cornell Medicine, New York, told Alzforum. “This is an unexpected aspect of tau pathology.” Gan plans to test whether this is true in other FTD mutations, and wonders about tau-induced hyperexcitability in Alzheimer’s disease, in which tau is not mutated.
“This study provides an important demonstration of the utility of human iPSC models to reveal changes in neuroplasticity that may lead to disruption of brain circuitry over the course of disease,” wrote Stephen Haggarty, Massachusetts General Hospital, Boston, to Alzforum. “[It] provides further validation of therapeutic strategies seeking to reduce expression of pathological forms of tau.”
Not all tau mutations work in this manner. In the September 19 Stem Cell Reports, researchers led by Hideyuki Okano of Keio University, Tokyo, report a different effect of the R406W. This mutation lies outside the microtubule-binding domain, but has nevertheless been reported to impede tau binding to those intracellular rails. Also using patient-derived neurons, first author Mari Nakamura and colleagues found that R406W tau mislocalizes to dendrites, as was seen before in cell and animal overexpression models (Thies and Mandelkow, 2007; Jan 2011 news). There it disrupts mitochondrial transport and causes axonal degeneration.
“In neither study do the cell models rely on overexpression of the mutant tau protein, thereby overcoming issues often inherent to artificially engineered systems,” Haggarty wrote.—Gwyneth Dickey Zakaib
References
Mutations Citations
News Citations
Paper Citations
- Rossi G, Tagliavini F. Frontotemporal lobar degeneration: old knowledge and new insight into the pathogenetic mechanisms of tau mutations. Front Aging Neurosci. 2015;7:192. Epub 2015 Oct 14 PubMed.
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998 Dec 4;282(5395):1914-7. PubMed.
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer's disease mouse model. Science. 2007 May 4;316(5825):750-4. PubMed.
- Beagle AJ, Darwish SM, Ranasinghe KG, La AL, Karageorgiou E, Vossel KA. Relative Incidence of Seizures and Myoclonus in Alzheimer's Disease, Dementia with Lewy Bodies, and Frontotemporal Dementia. J Alzheimers Dis. 2017;60(1):211-223. PubMed.
- Sayas CL, Tortosa E, Bollati F, Ramírez-Ríos S, Arnal I, Avila J. Tau regulates the localization and function of End-binding proteins 1 and 3 in developing neuronal cells. J Neurochem. 2015 Jun;133(5):653-67. Epub 2015 Apr 8 PubMed.
- Thies E, Mandelkow EM. Missorting of tau in neurons causes degeneration of synapses that can be rescued by the kinase MARK2/Par-1. J Neurosci. 2007 Mar 14;27(11):2896-907. PubMed.
Further Reading
Primary Papers
- Sohn PD, Huang CT, Yan R, Fan L, Tracy TE, Camargo CM, Montgomery KM, Arhar T, Mok SA, Freilich R, Baik J, He M, Gong S, Roberson ED, Karch CM, Gestwicki JE, Xu K, Kosik KS, Gan L. Pathogenic Tau Impairs Axon Initial Segment Plasticity and Excitability Homeostasis. Neuron. 2019 Nov 6;104(3):458-470.e5. Epub 2019 Sep 18 PubMed.
- Nakamura M, Shiozawa S, Tsuboi D, Amano M, Watanabe H, Maeda S, Kimura T, Yoshimatsu S, Kisa F, Karch CM, Miyasaka T, Takashima A, Sahara N, Hisanaga SI, Ikeuchi T, Kaibuchi K, Okano H. Pathological Progression Induced by the Frontotemporal Dementia-Associated R406W Tau Mutation in Patient-Derived iPSCs. Stem Cell Reports. 2019 Oct 8;13(4):684-699. Epub 2019 Sep 19 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.