Of Mice and Men—Functional Imaging of Aβ Toxicity, Early Pathology
Quick Links
Functional imaging may be a promising way to detect decline in Alzheimer’s and other neurodegenerative diseases, but can it detect those early synaptic deficits that are known to occur before neurons die? In the October Archives of Neurology, Scott Small and colleagues at the Taub Institute for Research on Alzheimer’s Disease and Columbia University, New York, report that functional magnetic resonance imaging (fMRI) detects similar deficits in both AD brain and AD mouse models that have no neuronal loss. “The study shows that fMRI is sensitive to cell sickness and not just cell death,” said Small in an interview with ARF.
That sentiment is echoed by Sterling Johnson and colleagues at the University of Wisconsin School of Medicine, Madison. Reporting in this month’s Archives of General Psychiatry, these researchers found that middle-aged, cognitively normal adults with a family history of AD show reduced brain activation in a self-appraisal test. This particular task challenges parts of the brain that are vulnerable in AD, including the hippocampus and the posterior cingulate. “Our data are entirely in keeping with the idea that deficits occur before cell death, since we did not find any brain atrophy in this study,” said Johnson. Together, the two studies support the idea that functional deficits can be detected, at least in specialized research settings, before neuronal loss.
Small and colleagues have spent 7 years devising a functional imaging platform that would give the same readout in mice and in humans. “That would greatly expand our experimental power,” he said. The requirements for such a platform are threefold. It should be sensitive to function, give high spatial resolution, and be applicable to both animal and human MRI. Of the four variables most commonly used for functional imaging—glucose metabolism, cerebral blood flow, deoxyhemoglobin, and cerebral blood volume—the researchers found that only the last fits the bill with current technology. The researchers have optimized this approach for primate and mouse brains, using, respectively, intravenous and intraperitoneal injections of gadolinium contrast agents. In the current study, first author Herman Moreno and colleagues applied the technique for the first time to humans and to J20 transgenic mice, which express human amyloid-β precursor protein (APP) with Swedish and Indiana mutations. As these mice age, they accumulate Aβ plaques but remain free of neurofibrillary tangles and neuronal loss (see Mucke et al., 2000).
Moreno and colleagues imaged 11 patients with probable AD and the same number of age-matched controls. The researchers obtained cerebral blood volume (CBV) maps of the whole brain, but paid particular attention to the hippocampus, the site of early degeneration in AD. Using anatomical criteria, Moreno and colleagues identified four hippocampal subregions: the entorhinal cortex (EC), dentate gyrus, CA1 subfield, and the subiculum. Analysis of variance showed that the AD patients had significantly reduced CBV in the entorhinal cortex; they also performed more poorly in cognitive tests, including hippocampal-dependent tests of memory.
Next the researchers turned to the mouse brains. Using the same technique, they imaged normal and transgenic mice at different ages to develop a timeline of functional decline. CBV in the entorhinal cortex, dentate gyrus, and the CA1 subfield were mapped (the subiculum gave unreliable signals). Again, the researchers found that reductions in the EC were dominant in the transgenic mice, beginning at about 6 months. Blood volume reductions spread throughout the hippocampus as the animals aged. Interestingly, the researchers were able to reduce EC blood volume loss in transgenic animals by treating them with R-flurbiprofen, a drug in phase 3 clinical testing. This enantiomer of the non-steroidal anti-inflammatory drug S-flurbiprofen is believed to modulate γ-secretase activity and reduce Aβ production.
“Taken together, our studies demonstrate that Aβ-related neurotoxicity in mice is sufficient to cause functional imaging defects as detected by CBV maps of the hippocampal formation,” write the authors. And, because they observed the same spatial pattern of defects in the human and mouse hippocampus (see figure below), they conclude that “the cross-species similarities support the likelihood that the patterns represent the same underlying Aβ-related pathophysiologic mechanism.” That implies that functional decline in humans can occur separately from both neuronal loss and neurofibrillary tangles (though see comment below from Richard Caselli). “This shows, for the first time really, that human functional imaging is sensitive to cell sickness,” said Small.
A man or a mouse
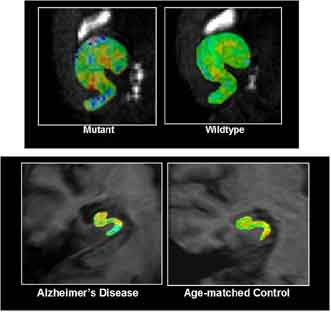
fMRI shows functional deficits in the hippocampus of APP mice (top left) compared to wild-type (top right). A similar pattern is seen when AD hippocampus (bottom left) is compared to normal (bottom right). Warmer colors indicate more activity. Image credit: Scott Small
Sterling Johnson and colleagues set out to answer a different question, but their findings are also consistent with the idea that functional deficits precede neuronal loss. “What we wanted to know was whether brain changes could be observed in people with risk factors for AD,” said Johnson. To test this, the researchers turned to BOLD, or blood-oxygenation level-dependent, fMRI, a technique that is now widely accepted as a faithful measurement of neuronal activity (see ARF related news story).
Johnson and colleagues measured BOLD signals while cognitively normal adults, aged 45-62, some with and others without a family history of AD, carried out a self-appraisal test. Volunteers were presented with “trait-adjectives,” such as “daring,” “controlling,” “sharp,” “friendly,” etc., and asked to decide with a simple yes or no if the word described them. The authors chose to look at self-appraisal for two reasons. The first is anatomical. “This task activates regions vulnerable to AD, including the hippocampus and posterior cingulate, which light up with PIB,” said Johnson. The second reason relates to the picture of AD pathology. “Everyone is focused on memory, and rightly so, but another problem with Alzheimer disease is with executive function, and metacognition could be considered a domain of executive function,” said Johnson. Self-appraisal, which involves reflective thinking about thinking, is a metacognitive function.
Johnson and colleagues found that in people with a family history (at least one parent with AD), the hippocampus and the medial parietal lobe was less active during the test. The finding supports an early study from this group that found people with a family history of AD to have fMRI deficits during a memory-encoding task (see Johnson et al., 2006). “Together, our papers suggest that risk for AD may alter brain function in people who are otherwise very cognitively normal,” said Johnson.
People who are cognitively impaired also show deficits in this test. Earlier this year, the researchers reported a significant correlation between BOLD signals and self-awareness in patients with mild cognitive impairment (see Ries et al., 2007). The researchers reasoned that patients with poor self-awareness might have reduced activity in brain areas that are activated during the self-appraisal task. “AD patients often have reduced insight into their own deficits,” said Johnson. Indeed, patients with the poorest cognitive abilities, as judged by comparing their own assessment of their cognitive skills with their spouse’s assessment, had the weakest fMRI signals.
Do the findings suggest that the researchers have detected signs of early pathology? “I think that is a strong possibility, but we would need to do more research to say for sure,” said Johnson. His group is planning to follow subjects longitudinally, testing them with the memory encoding paradigm. In fact, he is about to carry out a 4-year follow-up with the cohort and hopes to follow them until some might convert to AD, which could take 20 years, he said. Johnson also plans to image that cohort using PIB, which can identify Aβ plaques. Preliminary studies are underway.
Coming back to the question of deficits in the absence of neuronal loss, Johnson suggested that his group’s data are consistent with this idea, since voxel-based morphometry to measure grey matter volume found no differences between people with and without a family history of AD.
One surprising finding, suggested Johnson, is that when corrected for family history, ApoE genotype had no effect, despite the fact that it is the strongest risk factor to date for sporadic AD. “We think that there may be other genes, or gene environmental interactions, that we don’t have a good handle on yet,” he said. Interestingly, several PET imaging showed that ApoE4 carriers have reduced glucose metabolism in brain regions that are affected by AD, including the posterior cingulate (see Reiman et al., 2005). Johnson noted that those studies did not correct for family history, making it difficult to draw direct comparisons with his BOLD fMRI data. —Tom Fagan
References
Therapeutics Citations
News Citations
Paper Citations
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000 Jun 1;20(11):4050-8. PubMed.
- Johnson SC, Schmitz TW, Trivedi MA, Ries ML, Torgerson BM, Carlsson CM, Asthana S, Hermann BP, Sager MA. The influence of Alzheimer disease family history and apolipoprotein E epsilon4 on mesial temporal lobe activation. J Neurosci. 2006 May 31;26(22):6069-76. PubMed.
- Ries ML, Jabbar BM, Schmitz TW, Trivedi MA, Gleason CE, Carlsson CM, Rowley HA, Asthana S, Johnson SC. Anosognosia in mild cognitive impairment: Relationship to activation of cortical midline structures involved in self-appraisal. J Int Neuropsychol Soc. 2007 May;13(3):450-61. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A. 2005 Jun 7;102(23):8299-302. PubMed.
Other Citations
Further Reading
Papers
- Caselli RJ, Reiman EM, Locke DE, Hutton ML, Hentz JG, Hoffman-Snyder C, Woodruff BK, Alexander GE, Osborne D. Cognitive domain decline in healthy apolipoprotein E epsilon4 homozygotes before the diagnosis of mild cognitive impairment. Arch Neurol. 2007 Sep;64(9):1306-11. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proc Natl Acad Sci U S A. 2005 Jun 7;102(23):8299-302. PubMed.
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004 Jan 6;101(1):284-9. PubMed.
Primary Papers
- Johnson SC, Ries ML, Hess TM, Carlsson CM, Gleason CE, Alexander AL, Rowley HA, Asthana S, Sager MA. Effect of Alzheimer disease risk on brain function during self-appraisal in healthy middle-aged adults. Arch Gen Psychiatry. 2007 Oct;64(10):1163-71. PubMed.
- Moreno H, Wu WE, Lee T, Brickman A, Mayeux R, Brown TR, Small SA. Imaging the Abeta-related neurotoxicity of Alzheimer disease. Arch Neurol. 2007 Oct;64(10):1467-77. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
It is again interesting to see that individuals at presumably increased genetic risk (related to family history, but apparently not ApoE4) showed an altered fMRI-based activation pattern on a self-assessment task. This adds to a growing literature that presymptomatic "patients" have subtle but detectable alterations. I would have felt better, though, if the investigators had found this with ApoE4, because that is a known powerful genetic risk factor, and there is no evidence currently of a stronger one waiting to be discovered based on genome-wide SNP analyses.
View all comments by Richard CaselliThe major finding of Moreno et al. is that amyloid itself causes dysfunction in the absence of neuronal death or secondary tau aggregation, and that this dysfunction may be reversible with R-flurbiprofen (in the mice, anyway). This is provocative. I am not sure everyone will agree this proves quite yet that there was dysfunction in the absence of cell death. Unless I am misreading the paper, there was no pathology provided in the mice; instead this was inferred from previous studies of similarly aged animals. Likewise, the similarities between "mice and men" are used to imply that the amyloid dysfunction in the absence of death is operative in humans, too. This is possible, it’s interesting, and consistent with current thoughts about Aβ oligomers, and it’s a novel approach. But I do not think it can considered definitive at this point.
View all comments by Richard CaselliAlzforum
Author Q&A with Scott Small. Questions by Gabrielle Strobel.
Q: Brad Hyman's group showed years ago that people already have neuronal loss in the entorhinal cortex by the time they are diagnosed with AD (Gomez-Isla, 1996). How can you be sure that your 11 diagnosed AD patients have no neuronal loss in this area?
A: In fact, we are sure that the patients have an admixture of cell loss, “cell sickness,” amyloid plaques, and neurofibrillary tangles. The mouse studies are the ones that show that CBV maps are sensitive enough to capture Aβ-related “cell sickness” (because they never develop cell loss). Thus, we can conclude that CBV mapping and other functional imaging variables are, in principle, sensitive enough to capture the cell sickness stage of AD. This is important because as we apply CBV techniques to people with the earliest stage of disease, these findings suggest that we might be able to detect early pathophysiology, that is, cell sickness. Furthermore, from a therapeutic perspective, capturing this stage is important, since it is easier to treat a “sick cell” than a dead cell.
Q: In a nutshell, are you saying that while more is going on in the AD brain, the deficits your fMRI method visualizes are the piece that's due to Aβ synaptic toxicity?
A: In a nutshell, we are saying that we now know that functional imaging is sensitive, in principle, to the cell sickness stage of AD. Previous studies in humans could not conclude this because of the point you raise above.
Q: Floyd Bloom's group has characterized dendritic shrinkage in dentate gyrus early on in PDAPP mice (Wu et al., 2004) and hippocampal volume changes in 40-day-old PDAPP mice (Redwine et al., 2003). Have you imaged PDAPP mice to compare to the J20? How do these findings fit together?
A: Good question. Actually, Redwine et al. showed retraction of presynaptic terminals in the dentate. These presynaptic terminals “belong” to entorhinal cortex neurons. Thus, at face value, their findings and ours agree that entorhinal cortex neurons are involved. The observation that the mice had reduced volume in the dentate is interesting. I should point out that no one really knows what volume changes mean at a neurobiological level. Furthermore, from my conversations with Jeffrey Redwine about his paper, i take it they never measured volume of the entorhinal cortex. This would have been interesting. In any case, I agree applying this technique to different models would be interesting and is important. As we point out in the paper, over time we do see CBV changes in all hippocampal subregions in the mice. However, when collapsed over time, the entorhinal cortex changes are the dominant ones.
Q: Is your fMRI method of measuring the CBV variable, in mice and humans, highly specialized and bound to remain restricted to your academic center, or can it become widely available?
A: In principle, it does not need special scanners or software. It can be done anywhere. The analysis side is a little sophisticated but can be done anywhere, as well.
Q: I assume a goal is to make fMRI of CBV in entorhinal cortex a diagnostic tool of broad clinical value? What are the next steps in this direction?
A: This is one goal. There are others. Diagnostics, as you know, requires large numbers of subjects. Because of this I am pleased that we just finished acquiring CBV maps in hundreds of healthy elders. This is an NIA-funded project headed up by Richard Mayeux. Now, we will wait and see whether we can predict who will progress to AD and who will not. Time will tell. Other utilities are trying to understand mechanism (why is entorhinal cortex vulnerable to AD and why is dentate gyrus vulnerable to normal aging—this line of questioning gave rise to our retromer findings), and for monitoring drug efficacy.
Q: Do you think your method would make a better biomarker of drug response than, say, MRI volumetry or PIB PET, two imaging modalities that are gradually being incorporated in clinical trials for that purpose?
A: Unsure. This is an empirical question.
Q: Have you used the fMRI measure on people at risk, i.e., from two ApoE4 alleles, or a family history? Can you see the CBV change in them, too?
A: Yes.... Interesting findings.... This is a work in progress.
References:
Gomez-Isla T, West HL, Rebeck GW, Harr SD, Growdon JH, Locascio JJ, Perls TT, Lipsitz LA, Hyman BT. Clinical and pathological correlates of apolipoprotein E epsilon 4 in Alzheimer's disease. Ann Neurol. 1996 Jan;39(1):62-70. PubMed.
Wu CC, Chawla F, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Selective vulnerability of dentate granule cells prior to amyloid deposition in PDAPP mice: digital morphometric analyses. Proc Natl Acad Sci U S A. 2004 May 4;101(18):7141-6. PubMed.
Redwine JM, Kosofsky B, Jacobs RE, Games D, Reilly JF, Morrison JH, Young WG, Bloom FE. Dentate gyrus volume is reduced before onset of plaque formation in PDAPP mice: a magnetic resonance microscopy and stereologic analysis. Proc Natl Acad Sci U S A. 2003 Feb 4;100(3):1381-6. PubMed.
View all comments by Gabrielle Strobel�
Interesting study linking non memory dysfunction to early synaptic markers of pre-AD. But if self appraisal (self esteem ) is to be used as a valid trigger condition to evaluate functional blood flow in hippocampal, parietal associative-, and posterior cinguli cortex, then perhaps a third patient control group (non-familial AD, depressed) should be included. Depression has deleterious effects on self esteem. Also, depression is a documented risk factor in Alzheimer disease and is often found clinically together with symptoms of cognitive involution. It opens an interesting gateway between neurology and psychiatry.
Make a Comment
To make a comment you must login or register.