LRRK2 Variants Keep α-Synuclein from Forming Tetramers
Quick Links
More than a decade has passed since researchers led by Dennis Selkoe of Brigham and Women’s Hospital in Boston first reported that when α-synuclein has its druthers, it prefers the company of three others. Now, a new study led by Selkoe reports that disease-associated mutations in another PD gene, LRRK2, destabilize this tetrameric form of α-synuclein. Published September 16 in the journal Parkinson’s Disease, the study found that in indued neurons from LRRK2 mutation carriers who had PD, the ratio of α-synuclein tetramers to monomers was low compared to control cells, while phosphorylation of α-synuclein was high. Treating the cells with LRRK2 kinase inhibitors restored the tetramers and lessened the pathological phosphorylation. So did an inhibitor of stearoyl-CoA desaturase, an enzyme that nixes lipids thought to stabilize α-synuclein tetramers.
- PD-linked variants in LRRK2 reduced the proportion of α-synuclein in tetrameric form.
- The mutations also boosted α-synuclein phosphorylation at serine-129.
- LRRK2 kinase inhibitors, or an inhibitor of a fatty-acid-desaturating enzyme, prevented these effects.
“This is an interesting study, providing additional evidence on the potential pathogenic interplay between LRRK2 and α-synuclein,” wrote Huaibin Cai, NIA, Bethesda, Maryland. “The exact mechanisms of how LRRK2 modulates the oligomerization and phosphorylation of α-synuclein, however, remains to be determined.”
In 2011, Selkoe and colleagues caused a stir in the PD field when they reported that the most stable, aggregation-resistant, physiological form of α-synuclein is a tetramer (Aug 2011 news; Feb 2012 news). They later reported that disease-associated mutations in α-synuclein destabilized the tetrameric form, and that transgenic mice expressing a tetramer-reticent form of α-synuclein developed a PD-like disorder (Jun 2015 news). Most recently, researchers at Johns Hopkins University, Baltimore, extended these findings to mutations in glucocerebrosidase (GBA1), which also discouraged the formation of α-synuclein tetramers (Kim et al., 2018).
Might variants in LRRK2—after all, the most common genetic cause of PD—do the same? To investigate, first author Luis Fonseca-Ornelas and colleagues obtained induced pluripotent stem cells from four people with PD; two carried the G2019S LRRK2 variant, two had the R1441C variant. These mutations reside in the kinase and GTPase domains of this large protein, respectively. The scientists generated mutation-corrected versions of each iPSC line to serve as wild-type controls, then differentiated the iPSC lines into induced neurons for study.
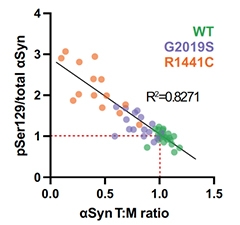
Fewer Tetramers, More Phosphorylation. In neurons induced from people carrying different PD mutations, the tetramer-to-monomer ratio of α-synuclein inversely correlated with the extent of phosphorylation at serine-129. [Courtesy of Fonseca-Ornelas et al., Parkinson’s Disease, 2022.]
Next, the scientists deployed cross-linking agents to stabilize existing multimers of α-synuclein within intact cells. They found that both LRRK2 mutations significantly dampened the ratio of tetramers to monomers. The LRRK2 variants also upped phosphorylation at serine-129, a hotspot for disease-associated hyperphosphorylation of α-synuclein. Across the cell lines, a negative correlation emerged, such that the more α-synuclein existed as a loner, the more of it was phosphorylated at serine-129.
Reasoning that revved-up kinase activity might be to blame for the effect of the LRRK2 variants, the researchers treated the cell lines with two inhibitors of the kinase, PF-06447475 and MLi-1, for several days while the neurons were maturing. Strikingly, either inhibitor prevented the dip in tetramerization caused by the mutations and also thwarted serine-129 phosphorylation.
“These results add to the collective body of data that LRRK2 kinase activity appears to (albeit subtly) push the normal biology of α-synuclein toward states associated with pathological conformations,” commented Andrew West of Duke University School of Medicine in Durham, North Carolina. “Next steps will be to determine how LRRK2 directs α-synuclein away from tetrameric states, whether acting directly, or indirectly through Rab phosphorylation or mitochondrial interactions, for example.”
Separately, the scientists also investigated how stearoyl-CoA desaturase inhibitors affect α-synuclein tetramerization. The SCD enzyme catalyzes the rate-limiting step in the production of monounsaturated fatty acids from saturated fatty acid precursors, and studies from Selkoe’s and other labs have shown that SCD inhibitors can relieve α-synuclein toxicity (Dec 2018 news). Recently, they also showed that an SCD inhibitor supported tetramer formation in transgenic mice that expressed tetramer-destabilizing form of α-synuclein (Oct 2020 news). Now, they report the same was true in the context of LRRK2 mutations. Although the SCD inhibitor did not influence LRRK2 kinase activity, it did restore the tetramer to monomer ratio in LRRK2 mutant neurons, and reduced the hyperphosphorylation of α-synuclein.
The researchers obtained similar results from the same series of experiments when they differentiated the patient iPSCs into dopaminergic neurons instead of cortical neurons.
How do mutations in LRRK2 discourage the formation of α-synuclein tetramers? Selkoe does not know, but he noted that mutations in α-synuclein itself, GBA1, and now, LRRK2, all seem to destabilize -synuclein tetramers, casting α-synuclein homeostasis as a central disease mechanism. That SCD inhibitors restore the tetramer-to-monomer ratio in LRRK2 mutant cells suggests that lipid metabolism can act as a downstream mediator of genetic insults, disrupting α-synuclein’s ability to oligomerize, Selkoe proposed.
Despite some converging evidence, not everyone is convinced that shifts in -αsynuclein multimerization are the central mechanism driving PD. “I don’t think we know at this stage whether loss of tetramers is damaging to neurons, although the association with dominant mutations of both LRRK2 and SNCA would imply so,” wrote Mark Cookson of the National Institutes of Health in Bethesda, Maryland. Cookson added that multiple studies suggest that LRRK2 plays an important role in lysosomal function in non-neuronal cells, which were not examined in the current study.—Jessica Shugart
References
News Citations
- An α-Synuclein Twist—Native Protein a Helical Tetramer
- Synuclein—Researchers Out of Sync on Structure
- Form and Function: What Makes α-Synuclein Toxic?
- Does Oleic Acid Hold the Key to α-Synuclein Toxicity?
- Curbing Fatty Acids Means No Parkinson’s—If You Are a Mouse
Paper Citations
- Kim S, Yun SP, Lee S, Umanah GE, Bandaru VV, Yin X, Rhee P, Karuppagounder SS, Kwon SH, Lee H, Mao X, Kim D, Pandey A, Lee G, Dawson VL, Dawson TM, Ko HS. GBA1 deficiency negatively affects physiological α-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A. 2018 Jan 23;115(4):798-803. Epub 2018 Jan 8 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Fonseca-Ornelas L, Stricker JM, Soriano-Cruz S, Weykopf B, Dettmer U, Muratore CR, Scherzer CR, Selkoe DJ. Parkinson-causing mutations in LRRK2 impair the physiological tetramerization of endogenous α-synuclein in human neurons. NPJ Parkinsons Dis. 2022 Sep 16;8(1):118. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
National Institute on Aging
This report pairs of IPSC neurons where LRRK2 mutation status associates with a shift in the tetramer-monomer ratio in neurons and an increase in pS129-synuclein. This latter observation is particularly interesting as the assay for measuring pS129-synuclein in neurons is robust and could be used across laboratories to validate the main results of the published work in multiple additional IPSC clones and presumably in knock-in mouse neurons. It may be of interest to note here that we have been working to make isogenic LRRK2 lines available as part of larger project to model multiple single variant causes of neurodegeneration (Human iPS Cells, The Jackson Lab).
Considering open questions that come from this report, it would be critical to further understand the mechanism(s) by which LRRK2 genotype affects synuclein metabolism. The application of two structurally distinct kinase inhibitors reversed all effects, including those on synuclein but also on Rab10 phosphorylation. However, it is curious that the authors used very extended and very high levels of inhibitors. Typically, measures of LRRK2 activity including pS1292 LRRK2 and Rab12 or Rab29 are responsive to inhibitors within one hour, including in vivo (Kluss et al., 2021). Additionally, pRAb10 is not very responsive to the LRRK2 G2019S mutation in some tissues (Iannotta et al., 2020; Fan et al., 2021).
These considerations may indicate that neuronal LRRK2, as evaluated here, may have different regulation than does LRRK2 in other contexts. Possibly relevant is that in non-neuronal cells, we, and several other labs, see a strong relationship between LRRK2 activity and lysosomal function (e.g., Bonet-Ponce et al., 2020) but work in neurons suggests a role of LRRK2 in autophagolysosome transport (Boecker et al., 2021). Thus, it would be important to understand whether synuclein is responsive to LRRK2 genotype in neurons because it is either not being degraded, which seems unlikely given that total synuclein levels are unchanged, or whether the distribution of protein between soma and synapse is altered.
The other unanswered question from this paper is whether an altered monomer-tetramer ratio, as evaluated by cross-linking then western blots, is responsible for any LRRK2 mutation effects. I don’t think we know at this stage whether loss of tetramers is damaging to neurons, although the association with dominant mutations of both LRRK2 and SNCA would imply so. It is notable that only a small amount of cross-linked material is recruited into the potential tetramers (Figure 2a in the current paper, for example). It would be interesting to repeat these experiments in vivo using LRRK2 knock-in (and knockout) alleles that have been endogenously introduced into the mouse genome. Unfortunately, at least in baseline conditions, those animals don’t have obvious TH cell loss so one would have to then challenge the animals further to be able to test relationships between tetrameric and phosphorylated synuclein, LRRK2 genotype, and dopamine cell loss. However, such experiments would then form the basis of reversal of both LRRK2 activation and synuclein tetramer formation; this could be dissected out, to some degree, using the SCD inhibitor reported by this group to reverse synuclein tetramer formation irrespective of LRRK2 genotype, presumably without affecting LRRK2 activity per se.
References:
Kluss JH, Mazza MC, Li Y, Manzoni C, Lewis PA, Cookson MR, Mamais A. Preclinical modeling of chronic inhibition of the Parkinson's disease associated kinase LRRK2 reveals altered function of the endolysosomal system in vivo. Mol Neurodegener. 2021 Mar 19;16(1):17. PubMed.
Iannotta L, Biosa A, Kluss JH, Tombesi G, Kaganovich A, Cogo S, Plotegher N, Civiero L, Lobbestael E, Baekelandt V, Cookson MR, Greggio E. Divergent Effects of G2019S and R1441C LRRK2 Mutations on LRRK2 and Rab10 Phosphorylations in Mouse Tissues. Cells. 2020 Oct 22;9(11) PubMed.
Fan Y, Nirujogi RS, Garrido A, Ruiz-Martínez J, Bergareche-Yarza A, Mondragón-Rezola E, Vinagre-Aragón A, Croitoru I, Gorostidi Pagola A, Paternain Markinez L, Alcalay R, Hickman RA, Düring J, Gomes S, Pratuseviciute N, Padmanabhan S, Valldeoriola F, Pérez Sisqués L, Malagelada C, Ximelis T, Molina Porcel L, Martí MJ, Tolosa E, Alessi DR, Sammler EM. R1441G but not G2019S mutation enhances LRRK2 mediated Rab10 phosphorylation in human peripheral blood neutrophils. Acta Neuropathol. 2021 Sep;142(3):475-494. Epub 2021 Jun 14 PubMed.
Bonet-Ponce L, Beilina A, Williamson CD, Lindberg E, Kluss JH, Saez-Atienzar S, Landeck N, Kumaran R, Mamais A, Bleck CK, Li Y, Cookson MR. LRRK2 mediates tubulation and vesicle sorting from lysosomes. Sci Adv. 2020 Nov;6(46) Print 2020 Nov PubMed.
Boecker CA, Goldsmith J, Dou D, Cajka GG, Holzbaur EL. Increased LRRK2 kinase activity alters neuronal autophagy by disrupting the axonal transport of autophagosomes. Curr Biol. 2021 May 24;31(10):2140-2154.e6. Epub 2021 Mar 24 PubMed.
These results add to the collective body of data that LRRK2 kinase activity appears to (albeit subtly) push the normal biology of α-synuclein toward states associated with pathological conformations.
Next steps will be determining how LRRK2 directs α-synuclein away from tetrameric states, whether acting directly, or indirectly through Rab phosphorylation or mitochondrial interactions, for example.
The new tools we have, including LRRK2 inhibitors that work at low nanomolar levels, and pT73-Rab10 antibodies that are quite effective for measuring LRRK2 kinase activity, build more confidence in the conclusions that are made.
Johns Hopkins University
This fascinating finding builds on the authors’ previous works highlighting an increase in the ratio of α-synuclein monomers and tetramers as a molecular mechanism driving PD. They show in this paper that LRRK2 mutations negatively affect the ratio of monomer to tetramer and increase phosphorylation at pSer129, effects that are rescued by an SCD inhibitor and a LRRK2 inhibitor. These results add to the collective body of data that genetic modifiers could affect the formation of tetramers of α-synuclein, and that changes in lipid homeostasis are an important mechanism in the formation and PD pathogenesis.
This is impressive work. That said, I think we need more information regarding:
1) the use of isogenic LRRK2 lines,
2) understanding the direct causal relationship between LRRK2 mutation and tetramer formation,
3) how LRRK2 mutation induces changes in lipid homeostasis,
4) SCD inhibition, how the accumulation of polysaturated lipids by LRRK2 mutation plays a role in the inhibition of tetramer formation-lipidomics data derived from cells with LRRK2 mutation would be informative for the answer,
5) whether the amount of specific lipids is important, or whether changes in the lipid composition of specific organelles or presynaptic terminal membrane are important.
Also, it would be important to understand whether α-synuclein 129 phosphorylation could inhibit tetramer formation.
INSERM
Fonseca-Ornelas and colleagues in Dennis Selkoe’s lab have used human induced pluripotent cells to study the relationships between LRRK2 and α-synuclein tetramers, which their lab has proposed in the past decade to be a “healthy” form of α-synuclein because it is resistant to pathological aggregation. The authors detect the tetramers using a cross-linking assay whereby they appear as different bands on western blots ranging from 60 to 100 kDa (compared to the 14 kDa monomer) and hence may be loosely described as lower-order complexes of α-syn. While previous work has reported that LRRK2 activity may be required for α-synuclein-mediated pathological events, such as cell death, α-synuclein phosphorylation, or cell-to-cell transmission of the protein, this is the first report that explores links between LRRK2 and these tetrameric species.
Assuming that such lower-order complexes are more desirable than α-syn monomers, the finding that the lower-order complex/monomer ratios are reduced in induced neuronal cells harboring pathological LRRK2 mutations suggests a potential pathway leading to adverse, long-term effects in these patients. Also, the observation that the lower-order complex/monomer ratio increased after treatment with LRRK2 kinase inhibitors further confirms that LRRK2 may be an important upstream regulator of α-syn toxicity.
Interestingly, the lower-order complex/monomer ratio also increased upon changes in cellular lipid composition, with conditions preventing the generation of monounsaturated fatty acids being favorable. This suggests that lipids are important in defining the equilibrium between α-synuclein monomer and its protective lower-order complexes. It remains to be verified if this is a direct effect of the lipids on α-synuclein that overrides the effects of LRRK2 mutants, or if the lipid composition in cells affects the functions of the LRRK2 mutants.
Should the described phenomenon be confirmed in larger cohorts of patients, this lower-order complex/monomer ratio may be interesting to use in stratifying patients for clinical trials or as a phenotype in therapeutic screening efforts.
Make a Comment
To make a comment you must login or register.