Losing NEMO: Could This Protein Explain COVID Brain Fog?
Quick Links
In some COVID-19 survivors, neurological problems linger, causing concern about when, and if, they will resolve and whether they might increase risk for dementia. What is behind those symptoms? Researchers led by Markus Schwaninger, University of Lübeck, Germany, charge the SARS-CoV-2 protease Mpro with killing brain endothelial cells, and in this way damaging the blood-brain barrier. In the October 21 Nature Neuroscience, the scientists reported more dead capillaries in brain tissue from people who had died from COVID-19 than from other causes.
- SARS-CoV-2 protease destroys endothelial transcription factor NEMO.
- This may explain dead brain capillaries during COVID.
- Blocking necroptosis kinase RIPK3 preserved virally infected vasculature.
In vitro, the viral protease cleaved NF-κB essential modulator. NEMO, a transcription factor, triggers host antiviral responses and prevents endothelial cell death. Without NEMO, endothelial cells perished by necroptosis. In NEMO knockout mice, inhibiting the necroptotic kinase RIPK3 preserved brain blood vessels and BBB integrity. Though preliminary, the findings suggest that blocking endothelial cell death via RIPK may help in severe COVID-19.
“This provides one of the few molecular mechanisms of action of SARS-CoV-2 on the brain and vasculature in general,” Costantino Iadecola, Weill Cornell Medical College, New York, told Alzforum. “I was struck how the authors connected autopsy pathology all the way to molecular biology confirmation in mice,” he added.
Stanley Perlman, University of Iowa, Iowa City, was unsurprised by these findings. “Endothelial cell damage is very consistent with what we think is going on and what other people have found [in COVID],” he said. Ditto for Thomas Wisniewski, New York University School of Medicine. “That BBB and endothelial cell damage can be an important mechanism for long COVID is consistent with past data and our own studies,” he told Alzforum.
Neurological symptoms, ranging from “brain fog” to stroke, plague some people who develop COVID-19, and can linger for months in survivors in a condition called long COVID (Harapan et al., 2021; Apr 2021 conference news; Groff et al., 2021; Taquet et al., 2021). Why neurological problems occur, or persist, remains unclear.
To cut through the haze, co-first authors Jan Wenzel, Josephine Lampe, and Helge Müller-Fielitz analyzed frontal cortex tissue from 17 people over 60 who had died from COVID-19, and 23 age- and sex-matched controls who had died from other causes, mostly cardiovascular, at the University Medical Centers in Hamburg-Eppendorf and Göttingen, Germany. Suspecting compromised blood vessels, the scientists immunostained them, looking for the endothelial marker CD34, vascular protein collagen IV, and the apoptosis marker caspase-3.
Compared to control tissue, COVID-19 tissue contained almost three times as many caspase-3-positive capillaries and 30 percent more “string vessels,” i.e., thin tubes of collagen lacking endothelial cells. These strings are a sign of dead capillaries (see image below). The differences held even after correcting for disease severity and comorbidities, which are more common among severe COVID patients. “The quality of the tissue samples solidly established increased string vessels as an autopsy finding in COVID,” Iadecola said.
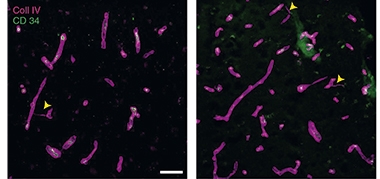
Ghosts of Capillaries Past. Compared to frontal cortex tissue from controls (left), tissue from people who had died from COVID-19 (right) contained more string vessels (arrowheads). [Courtesy of Wenzel et al., Nature Neuroscience, 2021.]
The researchers noticed that the COVID brain tissue resembled that of their mouse model of the rare genetic neurological disease incontinentia pigmenti, which is caused by a loss-of-function mutation in NEMO (Jiang et al., 2021). Other researchers had found that SARS-CoV-2 inhibits NEMO in cultured human lung epithelial cells (Wu et al., 2021). Could the same be happening in the brain? Indeed, after immunoblotting brain tissue lysates from four COVID cases and four controls, the authors saw more cleaved NEMO in two cases.
Suspecting a viral protease may be to blame, the scientists treated extracts from human or mouse endothelial or kidney cells that produced NEMO, with purified SARS-CoV-2 proteases. The virus makes two proteases, the papain-like protease and Mpro. Indeed, the latter cleaved NEMO. Further implicating Mpro, cultured human brain endothelial cells transfected with Mpro died, while cultures transfected with a mutated, inactive Mpro lived, with NEMO remaining intact (see image below).
Strung Out. Mice intravenously injected with the SARS-CoV-2 protease Mpro had more string vessels (arrowheads) in their cortices (middle) than did untreated mice (left) or mice injected with an inactive form of the protease (right). [Courtesy of Wenzel et al., Nature Neuroscience, 2021.]

To test the protease’s effect on NEMO in vivo, the researchers intravenously injected wild-type mice with a brain endothelial cell-specific viral vector carrying active or inactive Mpro. Two weeks later, the cortices of mice given the mutant protease were unaffected but, in mice given active Mpro, blood vessels were sparse and string vessels numerous—mimicking human COVID-19 tissue and brain tissue from mice infected with SARS-CoV-2. Mice with NEMO selectively knocked out of their brain endothelial cells also had more string vessels, and their BBB cell junctions were abnormally loose. The authors concluded that viral destruction of NEMO spelled the demise of brain endothelial cells.
But how did they die? NEMO blocks both apoptosis and necroptosis in endothelial cells. To figure out which was killing the cells, the scientists knocked out either the apoptosis signaling protein FADD, or the necroptosis signaling kinase RIPK3 in mice that were already lacking NEMO in their brain endothelial cells. Only the NEMO/RIPK3 double knockouts resisted brain damage. Ditto for RIPK3 knockout mice transfected with the Mpro protease (see image below). This indicates that necroptosis kills mouse endothelial cells lacking NEMO and that blocking this pathway can save them.
Better Without RIP. RIPK3 knockout mice (right) transfected with the SARS-CoV-2 protease Mpro had fewer string vessels (arrowheads) in their cortices than controls expressing the protease (left). [Courtesy of Wenzel et al., Nature Neuroscience, 2021.]

Could inhibitors of RIPK3, or other necroptosis proteins, treat COVID-induced vascular damage? As a proof of concept, the authors gave Mpro-expressing mice an experimental, brain-penetrating inhibitor of RIPK1, a kinase just upstream of RIPK3 in the necroptosis pathway. Collaborators at Sanofi supplied the compound. The treated mice formed no string vessels. The scientists are continuing to test the inhibitor in these mice, NEMO knockouts, and, eventually, SARS-CoV-2-infected mice.
Denali and Sanofi are testing the RIPK1 inhibitor DNL788/SAR443820, a successor to DNL747, in clinical trials for multiple sclerosis and amyotrophic lateral sclerosis (Zelic et al., 2021; Jun 2019 news). Last July, they began a Phase 1b trial of a non-CNS-penetrating RIPK1 inhibitor, DNL758/SAR443122, in people hospitalized with COVID-19 to tackle systemic hyperinflammation.
For the SARS-CoV-2 protease to cleave NEMO, it must be within endothelial cells. How does it slip inside? Scientists still debate whether SARS-CoV2 directly infects the brain. Only a few of the studies that looked for viral RNA or proteins within COVID brain tissue have found some (Jan 2021 news; Jun 2021 news). Likewise, Schwaninger's group found no expression of ACE2, the cell surface protein that SARS-CoV-2 uses as its portal into human cells, in brain endothelial cells. However, they did find high expression of two other possible receptors, BSG and NRP1, in a single-nuclei RNA sequencing dataset (Lake et al., 2018; Iadecola et al., 2020). This, along with viral RNA in endothelial cells from cerebral vessels of one COVID case, led the researchers to believe direct brain infection was possible.
Others are unconvinced. “This paper does not document viral persistence in the central nervous system, but only a possible tropism for the cerebral endothelium,” Tino Emanuele Poloni, Golgi-Cenci Foundation, Milan, Italy, wrote to Alzforum.
How else might the protease end up in cells? Iadecola thinks a leaky BBB may be to blame. “In the context of inflammatory disease, such as COVID-19, circulating pro-inflammatory cytokines can loosen the BBB and endothelial cell membranes, possibly allowing the viral proteins to enter,” he proposed.
While the researchers had no data about neurological symptoms in the postmortem cases in this study, Schwaninger said he does for a new set of cases and controls. He plans to see if memory deficits, seizures, and other neurological symptoms correlate with the number of string vessels.—Chelsea Weidman Burke
References
News Citations
- COVID-19 Worsens Neurological Problems, Delirium
- Dead Microglia Pave the Way for Myelin Regeneration
- How Does COVID-19 Affect the Brain?
- COVID-19 Prompts Choroid Plexus to Ring Alarm Bell
Therapeutics Citations
Paper Citations
- Harapan BN, Yoo HJ. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J Neurol. 2021 Sep;268(9):3059-3071. Epub 2021 Jan 23 PubMed.
- Groff D, Sun A, Ssentongo AE, Ba DM, Parsons N, Poudel GR, Lekoubou A, Oh JS, Ericson JE, Ssentongo P, Chinchilli VM. Short-term and Long-term Rates of Postacute Sequelae of SARS-CoV-2 Infection: A Systematic Review. JAMA Netw Open. 2021 Oct 1;4(10):e2128568. PubMed.
- Taquet M, Dercon Q, Luciano S, Geddes JR, Husain M, Harrison PJ. Incidence, co-occurrence, and evolution of long-COVID features: A 6-month retrospective cohort study of 273,618 survivors of COVID-19. PLoS Med. 2021 Sep;18(9):e1003773. Epub 2021 Sep 28 PubMed.
- Jiang Y, Müller K, Khan MA, Assmann JC, Lampe J, Kilau K, Richter M, Kleint M, Ridder DA, Hübner N, Schmidt-Supprian M, Wenzel J, Schwaninger M. Cerebral angiogenesis ameliorates pathological disorders in Nemo-deficient mice with small-vessel disease. J Cereb Blood Flow Metab. 2021 Feb;41(2):219-235. Epub 2020 Mar 9 PubMed.
- Wu J, Shi Y, Pan X, Wu S, Hou R, Zhang Y, Zhong T, Tang H, Du W, Wang L, Wo J, Mu J, Qiu Y, Yang K, Zhang LK, Ye BC, Qi N. SARS-CoV-2 ORF9b inhibits RIG-I-MAVS antiviral signaling by interrupting K63-linked ubiquitination of NEMO. Cell Rep. 2021 Feb 16;34(7):108761. Epub 2021 Feb 3 PubMed.
- Zelic M, Pontarelli F, Woodworth L, Zhu C, Mahan A, Ren Y, LaMorte M, Gruber R, Keane A, Loring P, Guo L, Xia TH, Zhang B, Orning P, Lien E, Degterev A, Hammond T, Ofengeim D. RIPK1 activation mediates neuroinflammation and disease progression in multiple sclerosis. Cell Rep. 2021 May 11;35(6):109112. PubMed.
- Lake BB, Chen S, Sos BC, Fan J, Kaeser GE, Yung YC, Duong TE, Gao D, Chun J, Kharchenko PV, Zhang K. Integrative single-cell analysis of transcriptional and epigenetic states in the human adult brain. Nat Biotechnol. 2018 Jan;36(1):70-80. Epub 2017 Dec 11 PubMed.
- Iadecola C, Anrather J, Kamel H. Effects of COVID-19 on the Nervous System. Cell. 2020 Oct 1;183(1):16-27.e1. Epub 2020 Aug 19 PubMed.
External Citations
Further Reading
Papers
- Favas TT, Dev P, Chaurasia RN, Chakravarty K, Mishra R, Joshi D, Mishra VN, Kumar A, Singh VK, Pandey M, Pathak A. Neurological manifestations of COVID-19: a systematic review and meta-analysis of proportions. Neurol Sci. 2020 Dec;41(12):3437-3470. Epub 2020 Oct 21 PubMed.
Primary Papers
- Wenzel J, Lampe J, Müller-Fielitz H, Schuster R, Zille M, Müller K, Krohn M, Körbelin J, Zhang L, Özorhan Ü, Neve V, Wagner JU, Bojkova D, Shumliakivska M, Jiang Y, Fähnrich A, Ott F, Sencio V, Robil C, Pfefferle S, Sauve F, Coêlho CF, Franz J, Spiecker F, Lembrich B, Binder S, Feller N, König P, Busch H, Collin L, Villaseñor R, Jöhren O, Altmeppen HC, Pasparakis M, Dimmeler S, Cinatl J, Püschel K, Zelic M, Ofengeim D, Stadelmann C, Trottein F, Nogueiras R, Hilgenfeld R, Glatzel M, Prevot V, Schwaninger M. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat Neurosci. 2021 Oct 21; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Gothenburg
CNS complications are common in acute severe COVID-19, and as SARS-CoV-2 infection also is linked to a substantial increased risk of thrombotic events, microvascular thromboembolic and hypoxic causes have been suggested as pathogenic mechanisms behind neurological complications in COVID-19.
Whether SARS-CoV-2 is neurotropic and directly infects cells in the brain is controversial and still not proven. During COVID-19 infection, SARS-CoV-2 is normally not detected in the cerebrospinal fluid, and pleocytosis (increased cell count) is normally absent, contrary to many other viral CNS infections.
In this impressive study, Markus Schwaninger and colleagues found indications of SARS-CoV-2 infection of endothelial cells in the brain and a possible link between infection and microvascular pathology by protease-mediated cleavage of NEMO, a major modulator of nuclear factor-κB. If these interesting findings hold, it may guide future research toward possible therapeutic interventions to treat neuropathology of COVID-19.
Golgi-Cenci Foundation & ASP Golgi-Redaelli
This elegant work illustrates a possible mechanism of neurological damage. However, the data reported does not document persistence of the virus within the central nervous system, but only a possible tropism for the cerebral endothelium documented in few cases. Indeed, as previously illustrated (Iadecola et al., 2020), the expression of ACE-2 in the brain appears quite low.
In my opinion, the "brain fog" phenomenon is not the result of a simple mechanism, such as direct SARS-CoV-2 invasion of the brain. Instead, it derives from a complex interaction between several biological (i.e., brain inflammation and hypoxia) and psychosocial factors (i.e., isolation and lockdown), which may accelerate neurodegeneration.
From a clinical point of view, a proper long-COVID definition, especially regarding the neuropsychiatric symptoms of what is commonly called brain fog, will be clarified through longitudinal studies that follow patients' courses. It should be considered that the recovery from COVID-19 requires termination of both viral infection and its associated inflammatory processes, which in severe cases takes much longer than four weeks.
Notwithstanding, some of the biological and psychosocial detrimental effects are at least partially reversible and "brain fog" can resolve.
References:
Iadecola C, Anrather J, Kamel H. Effects of COVID-19 on the Nervous System. Cell. 2020 Oct 1;183(1):16-27.e1. Epub 2020 Aug 19 PubMed.
Make a Comment
To make a comment you must login or register.