Lodged in Late Endosomes, Presenilin 2 Churns Out Intraneuronal Aβ
Quick Links
Presenilin 2 has long lived in the shadow of its sibling, presenilin 1. Either one can serve as the catalytic subunit of γ-secretase, but presenilin 1 has hogged much of the limelight, in part because many more pathogenic Alzheimer’s disease mutations are known to occur in this gene. But now, PS2 may have its own claim to infamy. According to a study published in Cell on June 9, presenilin 2 contains a sorting sequence that directs it to endolysosomal vesicles, where it produces intracellular Aβ peptides that are enriched for Aβ42. According to this new study, PS2’s highly specific localization stands in stark contrast to that of PS1. The latter calls many cellular compartments home, including the plasma membrane, from where its γ-secretase complex has long been known to release Aβ into the extracellular space. Wim Annaert at KU Leuven in Belgium led the study. Several commentators called the paper one of the most data-packed and methodologically complete they had read.
“There are very few papers as beautiful as this one,” commented Gunnar Gouras of Lund University in Sweden. “High-level research like this opens up new ways of thinking about what could be going wrong in AD.”
The findings may reinvigorate interest in intracellular Aβ, which has long been overlooked in favor of extracellular Aβ, commented Charles Glabe of the University of California, Irvine, who, like Gouras, was not involved in the study. Gouras, Glabe, and others have proposed that a pool of sticky intracellular Aβ kills neurons and ultimately seeds plaques (see Gouras et al., 2005; Gouras et al., 2010; and Pensalfini et al., 2014). Furthermore, PS2’s pivotal location underscores the central role of endolysosomal dysfunction in neurodegenerative disease, added Ralph Nixon of New York University, who proposed that changes in this endocytic system were among the earliest pathological events in sporadic AD (see Cataldo et al., 2000; Adamec et al., 2000).
The findings also distinguish the presenilins as more than just “proteasomes of the membrane” that chomp membrane proteins indiscriminately, as previously proposed (see Kopan and Ilgan, 2004). Rather, the spatial regulation of the proteases hints that they have specific physiological functions, the authors wrote.
Presenilin serves as the intramembrane protease within γ-secretase. The complex generates Aβ peptides from fragments of the amyloid precursor protein; it also cleaves many other substrates, including Notch. The lion’s share of familial AD mutations, more than 180, reside within PS1, while only 13 pathogenic mutations have been discovered in PS2. Gamma-secretase complexes containing PS1 or PS2 produce similar products in vitro, so researchers have tended to focus on PS1—generating mouse models and elucidating the structure of PS1-containing γ-secretase complexes. “I think the field has largely ignored possible differences in localization between the two presenilins, which could have a huge effect on substrate specificity,” Annaert said.
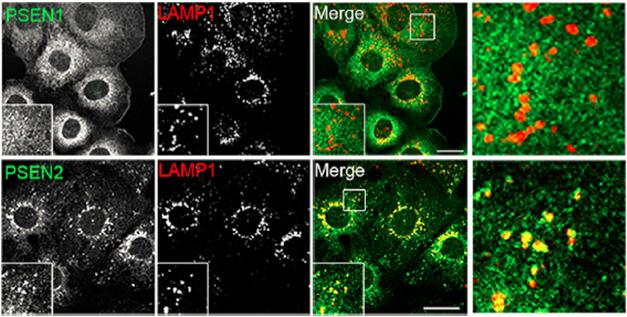
Endosome Home. PS2 (bottom), but not PS1 (top), resides in late endosomes and lysosomes, as evidenced by its co-localization with LAMP1 vesicles. [Courtesy of Sannerud et al., Cell 2016.]
First author Ragna Sannerud and colleagues set out to scrutinize presenilin localization and determine if compartmentalization within the cell would affect the respective complex’s proteolytic output. First examining mouse embryonic fibroblasts, they found that PS1 was spread across multiple cellular fractions, but nearly all PS2 was neatly restricted to late endosomal and lysosomal compartments (see image above). Nearly 10 percent of PS1 was detected in the plasma membrane on the cell surface, while only 1 percent of PS2 made it there. Confocal microscopy on several different cell lines and on mouse hippocampal neurons confirmed these findings, as PS2 comingled with vesicles positive for LAMP1, a marker of late endosomes and lysosomes. Rather than just relying on color co-localization to place PS2 in the cell, the researchers zoomed in even further, pairing structured illumination microscopy and correlative light electron microscopy to confirm that PS2 was indeed localized to membranes in these compartments (see image below). In neurons, these PS2-containing vesicles congregated in dendrites rather than axons or cell bodies.

Zooming in on PS2. Pairing structured illumination microscopy (left) and electron microscopy (right panels), researchers pinpointed PS2 (green) to the membranes of late endosomes, lysosomes, and multivesicular bodies (red). [Courtesy of Sannerud et al., Cell 2016.]
How were the presenilins sorted differently inside the cell? To find out, the researchers swapped different domains between PS1 and PS2. They found that the cytosolic N-terminus held the key to localization: Swapping this region shipped PS1 specifically to endolysosomes in fibroblasts and spread PS2 into membranes throughout the cell. The researchers ultimately homed in on a short sequence in PS2’s N terminus—E16RTSLM21. It fits the bill for an acidic dileucine motif that is common to other proteins sorted to endolysosomal compartments. Moreover, the researchers found that this sequence in PS2 bound to the cargo-sorting complex AP-1; mutating key residues in the dileucine motif broke this interaction and caused PS2 to be localized broadly. The association between AP-1 and PS2 was regulated by the phosphorylation of the serine-19 residue within E16RTSLM21, the researchers reported (see image below).

On Location. Starting from the trans-Golgi network, PS1 and PS2 take different paths in the cell. PS2 catches a ride with AP-1, which targets the protease to endolysosomal compartments that affect its Aβ output. PS1, on the other hand, spreads throughout the cell and onto the surface. Mutations in PS2’s sorting sequence alter the enzyme’s trafficking. [Courtesy of Sannerud et al., Cell 2016.]
Does PS2’s localization to the endosomal compartment affect the substrate specificity or activity of its respective γ-secretase complex? To find out, the researchers compared the processing of presenilin substrates N-cadherin, a cell surface protein, as well as Notch1 and APP, both of which are distributed throughout intracellular compartments and on the cell surface. For this the researchers used cells expressing either of the presenilins, isolating their membrane fractions. They found that PS2 processed N-cadherin less efficiently than PS1 did, and that, once again, swapping the sorting domains reversed this effect. Notch1 and APP were processed similarly by both presenilins to their respective intracellular fragments NICD and AICD.
What about Aβ production? The researchers found that cells expressing only PS2 contained higher amounts of intracellular Aβ and secreted less Aβ than did cells expressing only PS1. Regardless of which presenilin protein the cells expressed, the intracellular pool of Aβ had a higher ratio of Aβ42 to Aβ40 than did the secreted pool. While it is unclear why the intracellular pool would consist of longer Aβ peptides, Annaert proposed that something about this acidic compartment might affect the processing efficiency of the secretase. Alternatively, aggregation-prone Aβ42 could more strongly resist lysosomal degradation than shorter Aβ peptides can, and thus become overrepresented in that compartment, Nixon suggested.
Finally, the researchers investigated how familial AD mutations in PS1 and PS2 would affect both localization and functionality of the protein complexes. They found that none of four PS2 mutants—T122P, N141I, M239V, or M239I—altered its location. However, all of the mutations elevated the Aβ42/40 ratios, particularly in the intracellular pool. For the N141I mutant, for example, the intracellular ratio of Aβ42/40 shot up 50-fold compared to cells expressing wild-type PS2. The researchers concluded that the intracellular processing was particularly sensitive to the mutations.
Unexpectedly, the researchers found that two of five PS1 mutations, that is, L166P and G384A, acted essentially like phenocopies of PS2. They routed the complex to the endolysosomal compartment, giving it a PS2-like distribution. Cells expressing these PS1 mutants secreted less Aβ and racked up higher Aβ42/40 ratios than those expressing wild-type PS1.
It was striking that such a fundamental difference between PS1 and PS2 had gone unnoticed for more than 20 years of study, commented Todd Golde of the University of Florida in Gainesville. “This is an incredibly strong piece of work. It is important to encourage people not to just jump on the next new thing, but to remember there are still fundamental unanswered questions in the field.” He added that the importance of intracellular Aβ compared to its far more abundant extracellular forms had yet to be dissected.
Gabriele Kaminski Schierle of the University of Cambridge in England told Alzforum that the findings point to the importance of intracellular Aβ in the Alzheimer’s disease process, adding that her own work and that of others has revealed how readily Aβ clumps into aggregates when inside the cell (see May 2014 news). She wondered whether Aβ generated by PS2 in endolysosomes could go undegraded in people with AD. She proposed a cascade in which hyperactive neurons funnel Aβ-laden endosomes to the synapses, where their aggregated contents are released to neighboring cells. Alternatively, the vesicular Aβ buildup could trigger release of the contents into the cytosol, Schierle suggested. Glabe also mentioned the latter possibility, adding that the release of intracellular Aβ aggregates into the cytosol would trigger an inflammatory response.
“This study points to intracellular Aβ aggregation as a potential key point to pathogenesis in AD,” Glabe told Alzforum.
While Glabe believes the results support the notion that intracellular Aβ plays a central role in the endolysosomal dysfunction observed in AD, Nixon proposed it was the other way around: Waning endolysosomal function could precipitate AD before Aβ accumulates. “The paper raises the possibility that aggregation-prone Aβ could be a lysosomal toxin, but it could also be that undegraded proteins in general are bad for the cell,” he said. He proposed that age-related lysosomal dysfunction could trigger the buildup of many proteins, including intracellular Aβ produced by PS2. “This would broaden the relevance to sporadic AD, where we think multiple factors are driving the failure of the lysosome,” Nixon said.
What, if anything, does PS2’s specific role as an intracellular Aβ42 booster mean for antibody immunotherapy? Eric Siemers of Eli Lilly and Company in Indianapolis cautioned against drawing any conclusions in that regard. “Surprisingly little is known about subcellular compartmentation of γ-secretase and Aβ, so this is very interesting work. At this point, to try to relate their findings to the potential success or failure of monoclonal antibodies would be highly speculative, and probably not very useful in terms of moving the field forward,” he wrote to Alzforum. Lilly is on the verge of releasing results of its Expedition 3 trial on the Aβ immunotherapy solanezumab.
Gouras added that the intracellular pool might not escape therapeutic antibodies, as his work and others’ have revealed many antibodies are indeed taken up into neurons.—Jessica Shugart
References
Alzpedia Citations
Mutations Citations
News Citations
Therapeutics Citations
Paper Citations
- Gouras GK, Almeida CG, Takahashi RH. Intraneuronal Abeta accumulation and origin of plaques in Alzheimer's disease. Neurobiol Aging. 2005 Oct;26(9):1235-44. PubMed.
- Gouras GK, Tampellini D, Takahashi RH, Capetillo-Zarate E. Intraneuronal beta-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 2010 May;119(5):523-41. PubMed.
- Pensalfini A, Albay R 3rd, Rasool S, Wu JW, Hatami A, Arai H, Margol L, Milton S, Poon WW, Corrada MM, Kawas CH, Glabe CG. Intracellular amyloid and the neuronal origin of Alzheimer neuritic plaques. Neurobiol Dis. 2014 Nov;71:53-61. Epub 2014 Aug 1 PubMed.
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000 Jul;157(1):277-86. PubMed.
- Adamec E, Mohan PS, Cataldo AM, Vonsattel JP, Nixon RA. Up-regulation of the lysosomal system in experimental models of neuronal injury: implications for Alzheimer's disease. Neuroscience. 2000;100(3):663-75. PubMed.
- Kopan R, Ilagan MX. Gamma-secretase: proteasome of the membrane?. Nat Rev Mol Cell Biol. 2004 Jun;5(6):499-504. PubMed.
Further Reading
Papers
- De Strooper B, Annaert W. Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu Rev Cell Dev Biol. 2010 Nov 10;26:235-60. PubMed.
- Meckler X, Checler F. Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments. J Biol Chem. 2016 Jun 10;291(24):12821-37. Epub 2016 Apr 8 PubMed.
Primary Papers
- Sannerud R, Esselens C, Ejsmont P, Mattera R, Rochin L, Tharkeshwar AK, De Baets G, De Wever V, Habets R, Baert V, Vermeire W, Michiels C, Groot AJ, Wouters R, Dillen K, Vints K, Baatsen P, Munck S, Derua R, Waelkens E, Basi GS, Mercken M, Vooijs M, Bollen M, Schymkowitz J, Rousseau F, Bonifacino JS, Van Niel G, De Strooper B, Annaert W. Restricted Location of PSEN2/γ-Secretase Determines Substrate Specificity and Generates an Intracellular Aβ Pool. Cell. 2016 Jun 30;166(1):193-208. Epub 2016 Jun 9 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
RIKEN Center for Brain Science
A group led by Wim Annaert appears to have provided an answer to the question, “Why is the number of the pathogenic presenilin (PS) 2 mutations much smaller than that of PS1 mutations?” The authors demonstrate that PS2 is more tightly associated with acidic cellular compartments, i.e., late endosomes and lysosomes, than PS1, and that the acidic environments enable PS2/γ-secretase to produce more intracellular Aβ2 compared to Aβ40. PS2 therefore is already pathogenic in its wild-type state. The authors also demonstrate that the intracellular localization of γ-secretase activity affects their substrate specificity.
Some mysteries remain. Can the difference between PS1 and PS2 cellular localization, and thus substrate specificity, account for PS1 deficiency causing embryonic lethality, while PS2 deficiency does not? I used to attribute this to presumed weaker protein expression of PS2 than PS1 under normal conditions. If the antibodies to PS1 and PS2 used by the authors are of identical potency, the quantity of PS1 indeed overwhelms that of PS2 (Fig. 1A). The absolute quantities of PS1 and PS 2 need to be determined because the spatial information is only qualitative. This will also make clear whether wild-type PS2 is the cause of sporadic AD.
Another question concerns the etiological role(s) of intracellular Aβ42, the accumulation of which precedes extracellular deposition. I would like to humbly remind the authors and readers that it takes more than 20 years for Aβ amyloidosis to induce the disease symptoms (Bateman et al., 2012). There appear to exist a number of biological factors behind the chronic nature of AD. Some of them may proceed in hours, others in days, in months, in years, and in decades. This is why I call AD research “biology of time.”
References:
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
The University of Adelaide
I was curious to see that this paper did not address the previous observations of Area-Gomez et al. (2009) that a major site of subcellular localization of PSEN2 is the mitochondria-associated membranes of the ER, at least in mouse brain.
Since the autophagic pathway is required for Aβ secretion (Nilsson et al., 2013), accumulation of intracellular Aβ is to be expected from FAD mutations in the PSENs since such mutations (in PSEN1 at least) have been shown to decrease autophagy by reducing the function of the PSEN1 holoprotein in acidification of lysosomes (Lee et al., 2010). One might have expected a paper examining lysosomal localization of PSEN2 and intracellular Aβ accumulation to discuss the implications of Lee et al.
We should also remember that fAD mutations in PSEN2 are far fewer in number than those in PSEN1 and tend to show lower penetrance with a later average age of onset. If only a subset of fAD mutations in PSEN1 cause it to be localized in a similar manner to PSEN2, then the relevance of this subcellular localization to the pathological mechanism underlying Alzheimer’s disease is questionable. Differences in intracellular versus extracellular levels of Aβ40 vs Aβ42 accumulation can be predicted from the differential localization of PSEN1 and PSEN2 with respect to the ER as described by Area-Gomez et al. when integrated with the observations of Cook et al. (1997) regarding specific production of Aβ42 in the ER. See our recent review for further discussion (Jayne et al., 2016).
References:
Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, Yu WH, Duff KE, Yaffe MP, Pon LA, Schon EA. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009 Nov;175(5):1810-6. PubMed.
Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat Med. 1997 Sep;3(9):1021-3. PubMed.
Jayne T, Newman M, Verdile G, Sutherland G, Münch G, Musgrave I, Moussavi Nik SH, Lardelli M. Evidence For and Against a Pathogenic Role of Reduced γ-Secretase Activity in Familial Alzheimer's Disease. J Alzheimers Dis. 2016 Apr 4;52(3):781-99. PubMed.
Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013 Oct 17;5(1):61-9. PubMed.
Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, Uchiyama Y, Westaway D, Cuervo AM, Nixon RA. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010 Jun 25;141(7):1146-58. PubMed.
KULeuven & VIB
Takaomi Saido’s comment is right on the mark. It supports our statement that the field has overlooked important features associated with g-secretase heterogeneity that now can be addressed in more detail. To what extent our cell data address the issue of the small number of known PSEN2 FAD mutations is difficult to say. Maybe our geneticist colleagues can comment further on this.
I do agree with the idea that presenilin 2 might already be toxic in its wild-type state given its prominent role in intracellular Aβ generation. I would remain cautious, however, in stating that PSEN2 is causative for sporadic AD. That is because some PSEN1 FAD mutations phenocopy PSEN2 FAD with respect to localization, effects on intracellular Aβ, and substrate specificity. Rather, I would rephrase this into a focus on the pathological relevance of intracellular Aβ.
With respect to the observed differences in embryonic development, I suspect differences in absolute protein expression between PSEN1 and PSEN2 contribute to it but are not the determining factor. First, one does not need a lot of γ-secretase activity to have normal processing, and relative expression levels of both PSENs can differ a lot depending on the cell type. We found several examples of cell lines (though I’ll admit these are cancer cell lines), where relative expression of PSEN2 is equal to or even higher than that of PSEN1.
Second, and I think more importantly, there are two other aspects that may account for the observed developmental phenotypes. If one looks at the expression profile from embryonic stages to adulthood, PSEN2 expression takes off later than PSEN1 expression. PSEN2 expression peaks shortly after birth, whereas PSEN1 expression is already high at early embryonal stages and then declines. Given that development depends on optimal signaling pathways such as those mediated through Notch, PSEN1 deficiency is likely to more dramatically affect embryonic development and PSEN2 cannot compensate as it is expressed somewhat later.
Vice versa, given its earlier higher expression, PSEN1 is more likely to compensate for the loss of PSEN2 in early embryonic development.
On top of that, PSEN2’s more restricted distribution in late endosomes/lysosomes makes it less accessible to a broader range of substrates relevant for development, in particular those that are preferentially located and restricted to the cell surface. This further decreases the ability of PSEN2 to compensate for a loss of PSEN1 expression. Hence, it is not necessarily the absolute amounts, but rather expression profile combined with localization that may explain the dramatic differences between PSEN1 and PSEN2 deficiency.
I endorse the statement that AD research can be called “biology of time.” If I were to translate it to our daily experiments, it’s a call to be more cautious about classical AD transgene models and cellular overexpression approaches. They surely allow us to recapitulate major aspects of the pathological process, but they largely fail to help us understand the aging effects on disease onset and pathogenesis. These occur over such long periods of time, at endogenous levels of expression and with a multifactorial nature.
With regard to Michael Lardelli’s comment on ER-localized Aβ, we do not think that this majorly contributes since our PSEN2 transport mutants clearly demonstrate that the size of the intracellular pool dynamically and majorly changes with altering the localization of PSEN2. Moreover, if present, ER-localized Aβ is in the biosynthetic/secretory pathway, allowing it to more readily exit the cell as opposed to LE/LYS Aβ. We have not yet investigated the localization of PSEN2 in ER-mito contacts, but now have the GFP-tagged cell lines available to image this at more physiological concentrations and at a resolution that allows us to resolve such contacts. After all, much of these data are provided with overexpressed PSEN2, of which we know that it gets jammed in the ER overloading these compartments. The reported role of PSEN1 in lysosomal acidification may be a γ-secretase independent finding caused by PSEN1 deficiency, and as such is not directly part of our work on differential localization.
Institut de Pharmacologie Moléculaire et Cellulaire
The study by Sannerud and colleagues is a huge piece of work. By combining state-of-the-art cell biology techniques, it nicely delineates the specific structural properties of presenilin 1 or presenilin 2 that govern their subcellular localization and thereby, substrate specificity. The demonstration that PS2 occurs mainly in late endosome/lysosome compartments where intracellular Aβ accumulates is of utmost importance.
The fact that pathogenic mutations in PS1 alter the localization of PS1 by mimicking the localization of PS2 is also fascinating. This data suggest that the functional importance of PS2 in Alzheimer pathology has likely been underestimated, mainly because PS knockout experiments and associated in utero lethality suggested that PS1 was the main contributor to Aβ production.
The main unanswered question that remains is: What is the role of wild-type PS1 in sporadic cases of Alzheimer’s disease? Does it reflect an additional catabolic pathway on APP or other substrates that could contribute to the pathology?
We recently published an article where, for the first time, we combined bidirectional inducible promoter and 2A peptide technology to express the six possible combinations of the four γ-secretase components in a timely and stochiometric manner (Meckler and Checler, 2016). We showed in this article that the subunit composition of the γ-secretase complex governs distinct levels of its maturation and function, thereby differently modulating Aβ secretion. Interestingly, we also documented a PS-specific subcellular localization of the γ-secretase complexes, with PS1-containing complexes occurring at the plasma membrane and PS2-containing complexes targeted to the trans-Golgi network and late endosomes.
Furthermore, constructs we generated allowed us to establish the influence of a given Aph1 variant and to unravel that PS2-containing γ-secretase complexes are targeted to distinct endosomal compartments in an Aph1 subunit-dependent manner. This increases the level of complexity related to γ-secretase localization, suggesting it not only depends on the nature of the presenilin but is also linked to the Aph1 variant incorporated within γ-secretase complexes.
We do believe that constructs we generated would substantially help us to better understand the many aspects of γ-secretase localization and function. It would be particularly informative to assess the influence of pathogenic mutations harbored by the six distinct complexes. We are obviously ready to make these tools available to members of the scientific community who are interested in this field.
References:
Meckler X, Checler F. Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments. J Biol Chem. 2016 Jun 10;291(24):12821-37. Epub 2016 Apr 8 PubMed.
Massachusetts General Hospital
In response to Drs. Saido and Annaert, it is interesting to consider why there are so many more pathogenic early onset familial Alzheimer’s disease mutations in PSEN1 than in PSEN2. The numbers of mutations arising in one gene versus another depends on two main factors. First, one must ask whether the genomic region is more prone to mutation; this can be assessed by recombination rate. Second, one can ask how mutations that arise in a given gene affect survival of the carrier and their reproductive fitness. The recombination rates for PSEN1 and PSEN2 are similar, 1.3 cM/Mb and 1.0 cM/Mb, respectively, suggesting that the occurrence of new mutations should be roughly equal for both genes.
This leaves the question of whether there was more natural selection for PSEN1 versus PSEN2 mutations in evolution. The first consideration is whether PSEN1 mutations were more tolerable than those in PSEN2; the second, whether PSEN1 mutations perhaps conferred an evolutionary advantage over those in PSEN2. This can be considered in the context of γ-secretase activity in general, or with respect specifically to Aβ. With regard to Aβ, the new data from Sannerud et al. raise the question whether accumulation of intraneuronal Aβ due to PSEN2 mutations is less advantageous, or detrimental to survival, than increased deposition of extracellular Aβ due to PSEN1 mutations. Arguing that detrimental effects of these mutations were negatively selected against in evolution does not work well, since the effects on brain function occur post-reproductively.
This leaves the provocative question of whether PSEN1 vs PSEN2 mutations at one time in the evolution of humans may have conferred a selective advantage, e.g., with regard to their differential expressions in the body and effects on Aβ deposition. At the risk of oversimplification, with regard to Aβ, one might distinguish between mutations in PSEN1 versus PSEN2 as the former mainly promoting extracellular deposition and the latter mainly increasing intracellular Aβ accumulation. Given our recent study showing that Aβ functions as an anti-microbial peptide to fight viral, bacterial, or fungal infection as oligomers or fibrillar deposits, we can ask whether early-onset FAD mutations in PSEN1 could at one point in evolution have conferred a selective advantage to survival in reproductive years.
As an exercise in speculation, let’s consider ancestral human populations with lifespans around 30 years, generally reproducing soon after puberty. If early onset FAD mutations conferred protection against outbreaks of meningitis, encephalitis, or other brain infections (as shown in Kumar et al., 2016, in 5XFAD mice), by constitutively depositing extracellular Aβ starting as early as in the womb, then one could imagine at least one theoretical scenario in which there could have been positive selection for EO-FAD mutations in evolution. Continuing that same line of speculation, young adults with EO-FAD mutations would have survived meningitis and encephalitis outbreaks better than non-carriers, and gone onto reproduce, versus non-carriers.
References:
Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. 2016 May 25;8(340):340ra72. PubMed.
RIKEN Center for Brain Science
If the quantity of PS2 is much smaller than that of PS1 throughout development and aging, this will explain essentially every question about the difference between PS1 and PS2, and differential localization of PS2 may then be less important. Again, I urge γ-scretase researchers to determine the absolute quantities of these proteins. Immunochemical detection is qualitative because the signal intensity depends on the specificity and avidity of antibodies used, and the quantity of mRNA does not always correlate with the quantity of protein. One strategy would be to generate knock-in mice, in which a tag sequence is added at the N-termini.
National Institute of Neuroscience, NCNP
The study by Sannerud et al. presented clear evidence that PS1 and PS2 are differentially localized, with PS2 being preferentially present in late endosomes/lysosomes. It is an impressive finding that the interaction of AP-1 with PS2, but not PS1, plays a critical role in determining the subcellular locations of PS2. Consistent with this finding, Checler and colleagues (Meckler and Checler, 2016) demonstrated similar data on the differential locations of PS1- and PS2-containing γ-secretase complexes, describing a little bit different locations of PS2-complexes.
The finding that familial Alzheimer’s disease-associated mutations of PS2 and some of FAD-PS1 mutations dramatically increase intracellular Aβ42/40 ratios underscores the importance of intracellular Aβ42 in AD pathophysiology. The data are highly consistent with our previous work, which analyzed intracellular Aβ40 and Aβ42 in neuroblastoma cells co-expressing APP and PS1 (wild-type or G384A) or PS2 (wild-type or N141I) (Takeda et al., 2004). However, it should be considered that the clinical phenotypes of patients with PS2 mutations are generally milder than those with PS1 mutations. Further research is required to clarify the contribution of intracellular Aβ42 to the pathological process of AD.
References:
Meckler X, Checler F. Presenilin 1 and Presenilin 2 Target γ-Secretase Complexes to Distinct Cellular Compartments. J Biol Chem. 2016 Jun 10;291(24):12821-37. Epub 2016 Apr 8 PubMed.
Takeda K, Araki W, Tabira T. Enhanced generation of intracellular Abeta42 amyloid peptide by mutation of presenilins PS1 and PS2. Eur J Neurosci. 2004 Jan;19(2):258-264. PubMed.
Make a Comment
To make a comment you must login or register.