LOAD of Data Place Vascular Malfunction as Earliest Event in Alzheimer’s
Quick Links
Updated July 13 to correct apolipoprotein nomenclature.
By the time late-onset Alzheimer’s disease emerges from the shadows, the tangled web of biological intrigue that caused the disease is nearly impossible to tease apart. Now, in a study incorporating a hefty amount of imaging and biomarker data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI), researchers present AD as a pathological chain of events tripped off by malfunctions in the brain’s vasculature. Based on a data-driven model of disease progression spanning 30 years, they claim that cerebral blood flow wanes first, then amyloid builds up, brain metabolism declines, and neurons eventually die. The researchers, led by Alan Evans of the Montreal Neurological Institute in Quebec, also contend that fluid biomarkers of vascular, immune, and metabolic dysfunction appeared more abnormal than fluid markers classically associated with AD pathology, such as CSF Aβ42 and tau. Many scientists questioned the authors’ novel methodology, wondering if it distorted the results. Others were impressed by the scope of the analysis and think the findings, published June 25 in Nature Communications, bolster the hypothesis that vascular dysfunction occurs in the earliest stages of AD.
Berislav Zlokovic of the University of Southern California in Los Angeles said the study confirms the prominence of vascular dysregulation in AD. “Given that the findings are from one of the largest and most comprehensive biomarker analyses ever conducted, it is probably a good time for us to begin thinking about how to update our model of the Alzheimer’s pathophysiological cascade by including vascular dysfunction as a driver of disease pathogenesis.”

Vicious Vessels. Vascular abnormalities appeared first and remained more prominent than any other biological factor throughout AD progression. [Image courtesy of Iturria-Medina et al., Nature Communications 2016.]
Efforts to detect and treat AD as early as possible necessitate understanding how and when the disease starts. This is a tall order, because the disease process may begin two decades or more prior to the first clinical symptoms. During that time multiple factors, including genetic variation, cardiovascular disease, and metabolic disorders such as diabetes, may influence the onset of amyloid and tau pathology, and researchers debate which most likely trigger the disease. Risk factors may also differ significantly within populations, and multiple factors might be at work in the same person.
While many studies have modeled the progression of AD, none have yet included as many potential disease correlates at once. The most commonly cited staging diagram—devised by Clifford Jack of the Mayo Clinic in Rochester, Minnesota—overlays individual progression curves for amyloid-PET, CSF Aβ42 and tau, cognitive impairment, and brain glucose metabolism, showing each biological factor as worsening over time in a consecutive manner (see image below). It does not track progression of vascular dysregulation or functional impairment by fMRI, which many consider a more direct measure of neuronal function than FDG-PET measures of glucose utilization (see Feb 2013 conference coverage). Rather, it considers vascular dysregulation as one of several comorbidities that may hasten the onset of clinical symptoms in people who already harbor latent disease, according to the revised model presented in the 2013 paper (see Jack et al., 2013).
Furthermore, the Jack and other progression models are hypothetical, based on data from multiple studies, as opposed to a model that tracks progression of multiple factors in the same cohort. Evans’ group previously published a data-driven model of regional amyloid pathology in the brain using ADNI data, which suggested that Aβ spread quickly throughout connected regions (see Nov 2014 news). However, Evans pointed out that amyloid is but a single factor in AD progression, and ADNI data on multiple factors are ripe for the picking.
First author Yasser Iturria-Medina and colleagues drew from ADNI’s massive resource of imaging, fluid biomarker, and clinical data to chart changes in multiple biological factors across LOAD progression in 40- to 70-year-olds. First, the researchers used 7,700 brain images of 1,171 people to track changes in five biological factors. In healthy controls as well as people with diagnoses ranging from early mild cognitive impairment (EMCI) to late cognitive impairment (LMCI) to LOAD, researchers used arterial spin labeling to measure cerebral blood flow, florbetapir-PET to analyze amyloid deposition, FDG-PET as a measure of brain glucose metabolism, functional magnetic resonance imaging (fMRI) to look at neuronal function, and structural MRI to watch for brain atrophy. The researchers took stock of each of these measurements in 78 regions spanning the entirety of the brain’s gray matter. The measurements drew from a combination of cross-sectional and longitudinal data, as the number of follow-up measurements varied between people and imaging modalities in the heterogeneous ADNI data set.
For each imaging modality, signal values were fitted on a scale between -1 and 1, around a mean of zero. Then, for each age, disease stage, and brain region, the researchers developed an “abnormality value” based on how much the signal differed from that in healthy controls. They could then trace and compare these abnormality values, which lie between 0 and 1, to one another. Cerebral blood flow crossed into the abnormal zone first, and it remained the most abnormal factor throughout the course of the disease. This vascular component was 80 percent more abnormal across all brain regions and disease stages than any other biological factor. Some researchers cautioned that presenting the data as relative values this way may inflate small effects.
Amyloid deposition came next in terms of how abnormal it was, followed by metabolic dysfunction, functional impairment, and gray matter atrophy. Interestingly, when the researchers added cognitive data and classical AD biomarker data to their analysis, they found that memory problems occurred early in the disease process when vascular dysregulation was obvious and amyloid deposition, metabolic and functional impairments were beginning to crop up, but prior to the detection of abnormalities in CSF Aβ42, tau, or p-tau. They concluded that cognitive decline in LOAD is not the culmination of many brain changes, but rather it manifests even when early abnormalities, such as vascular dysregulation, are the only ones in play. To Evans and colleagues, the findings also indicate that the classical CSF biomarkers associated with AD are not the earliest red flags for disease, a conclusion that contradicts previous studies and also the Jack progression curves.
Evans told Alzforum he was surprised by this finding, but that it was rigorously scrutinized and not simply a product of the authors’ abnormality index technique. Rather, he said the large number of subjects included in the study allowed the researchers to detect small effect sizes in memory and other biological factors that smaller studies would not have picked up.
“Their approach is a tour de force in the field of progression tracking,” commented Ashish Raj of Weill Cornell Medical College in New York. “Although others have used similar techniques for aligning biomarkers along a common age axis, this study stands out in the amount of data sets and biomarkers used.”
Other researchers viewed the results with skepticism, however. David Holtzman of Washington University in St. Louis pointed out that other longitudinal and cross-sectional studies have reported that CSF Aβ42 begins to drop prior to the detection of amyloid on PiB scans, which are more sensitive than the florbetapir scans used in this study. He also noted that the ADNI dataset is skewed toward older, more impaired people. “I am very concerned about the conclusion drawn from this data, namely that vascular dysregulation is the earliest event leading to LOAD,” Holtzman wrote (see comment below). Anne Fagan, also of Wash U, commented that while subtle changes in episodic memory are known to occur in the preclinical phase of disease, the finding that such changes occur before all other pathological measures does not agree with other studies, and may reflect the authors’ use of abnormality indices.
In a separate abnormality analysis, the researchers examined changes in the concentrations of 87 CSF and 146 plasma protein biomarkers. In the CSF, they found that heart type fatty acid binding protein (hFABP) stood out as the most abnormal marker. This protein has been linked to LOAD progression and neurodegeneration before, and also serves as a marker for cardiovascular disorders (see Alzbiomarker; Chiasserini et al., 2010; Guo et al., 2013; and Ghani et al., 2000). Cortisol and apolipoprotein (a)—two proteins also associated with cardiovascular malfunctions—were next in line (see Toledo et al., 2012; Erqou et al., 2010). The classical AD biomarkers tau, p-tau, and Aβ42 ranked fourth, sixth, and 13th, respectively. The researchers suggested that hFABP, Apo(a), and cortisol could serve as biomarkers to include in any study seeking to detect AD at its earliest stages.
In plasma, interferon-γ-induced protein (IP-10) ranked most abnormal. This protein is a marker for peripheral inflammation and also regulates angiogenesis (see Oxenkrug 2011; Bodnar et al., 2006). Pregnancy-associated plasma protein A (PAPP-A)—a predictor of cardiovascular distress including heart attack—ranked next (see Li et al., 2013). Proinsulin, the precursor to insulin, ranked third in abnormality, hinting at a connection with metabolic dysfunction.
Henrik Zetterberg of the University of Gothenburg in Sweden wondered whether the new biomarker findings were a product of the researchers’ use of abnormality indices, as well. He questioned whether the technique could have artificially expanded differences between controls and diseased individuals. Several researchers, including Zetterberg, Fagan, and Holtzman, said the statistical methods used in the paper were not sufficiently understandable, and that it was unclear based on the methods section alone exactly how abnormality was determined.
One other potential caveat is that reportedly, a quarter of ADNI participants with MCI are amyloid-negative (see Aug 2013 conference coverage). This could mean they were not developing AD, and instead could harbor any number of other cognition-damaging pathologies, including vascular dementia, hippocampal sclerosis, Lewy body disease, or frontotemporal lobar degeneration. While Evans and colleagues did remove outliers based on spurious results on cognitive tests, they did not specifically remove people who had no signs of amyloid deposition, Iturria-Medina told Alzforum. Holtzman and others cautioned that this could skew the results away from amyloid prominence in AD. However, Iturria-Medina contends that the binary separation of subjects based on amyloid positivity is a flawed concept that prevents a deeper understanding of the causal mechanisms underlying disease.
The authors took care to point out that their progression model does not prove that vascular dysfunction is the root cause of AD; however, the fact that it precedes amyloid deposition does underscore its importance in the disease process, Evans insists. The findings support the idea that Aβ accumulates in the brain due to insufficient clearance, rather than Aβ overproduction, the researchers wrote.
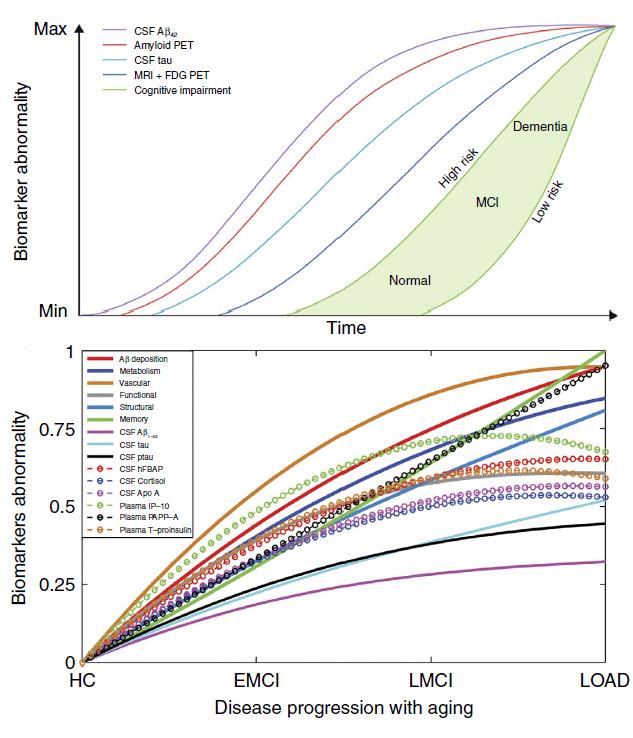
Cacophony of Curves. A comparison of the popular progression model proposed by Jack et al. (top) and a new model using ADNI data. [Image courtesy of Iturria-Medina et al., Nature Communications 2016.]
Alex Roher of Banner Sun Health Research Institute, Sun City, Arizona, praised the study for its thorough and unbiased approach. “They put all the cards on the table, and then to the surprise of many people in the field, they found that the most important marker of AD progression is cardiovascular dysfunction,” Roher said. While the progression model cannot prove this definitively, Roher said the results were highly suggestive that vascular problems precede, and likely cause, the deposition of amyloid. “A magic wand doesn’t cause amyloid to build up,” Roher said. “If you have a protein that accumulates, it does so for a reason.”
The next step will be to determine which vascular problems—hypertension, atherosclerosis, or others—cause the disease, Roher said. He added that a thorough cardiovascular work-up should become a part of any clinical exam regarding AD, and that more research should focus on systemic causes of AD, rather than a singular focus on the accumulation of amyloid. Hypertension is an established risk factor for LOAD.
Costantino Iadecola of Weill Cornell Medical College in New York said the study confirms myriad observational and animal studies that have pointed to the prominence of vascular dysfunction in AD. “This study put it all together in the same patients,” Iadecola said. “The power of that is tremendous.” However, he cautioned against drawing a causal relationship between vascular problems and amyloid deposition based on these results alone. He added that while vascular abnormalities preceded amyloid-PET scan abnormalities, it is impossible to know whether toxic amyloid oligomers, which are not detected on the scans, may have been present prior to the detection of plaques. “Now we know that plaques are more of a tombstone rather than where the action is,” he said. “The cognitive problems the researchers detected early in the disease may therefore be due to amyloid oligomers, vascular problems, or both.”
Evans acknowledged this could be the case, and said that his lab is in the process of developing a directional, causal model that could sort out these relationships. However, he added that the fact that memory problems and vascular dysregulation were apparent prior to abnormalities in CSF Aβ42 further suggests that vascular issues precede amyloid accumulation.
Roy Weller of the University of Southampton in England called the study a significant step forward in understanding AD progression. He added that while Aβ may accumulate due to insufficient clearance by a faltering vascular system, more attention should be given to other routes of clearance. “In assessing the role of arteries and capillaries in the brain, the perivascular route for elimination of fluid and solutes including Aβ should not be ignored,” he wrote. Experimental studies have shown that perivascular elimination of fluid and solutes is impaired with increasing age and this is reflected in the accumulation of Aβ in the walls of capillaries and arteries as cerebral amyloid angiopathy in Alzheimer’s disease (see Weller et al., 2015; Bakker et al., 2016).
Evans also mentioned the importance of the recently discovered glymphatic system, which drains solutes from the brain (see Aug 2012 news; Illif et al., 2013). However, he added that the efficiency of Aβ drainage is intimately linked to the pulsation of arteries in the brain. Therefore, vascular problems such as atherosclerosis would compromise the clearance of Aβ and other solutes from the blood vessels as well as glymphatics, he said.
All in all, Evans said the take-home message from this giant data analysis should be that AD is a multifactorial disease. While the vascular system may play a starring role, the exact causes of cognitive decline may differ between people depending on a slew of environmental, genetic, and lifestyle factors.—Jessica Shugart
References
News Citations
- HAI—Sharper Curves: Revamping a Biomarker Staging Model
- The Epidemic in Your Head? New Model Casts Amyloid as Intra-Brain Contagion
- Suspected Non-Amyloid Pathology (SNAP)—Not an Open or Shut Case
- Brain Drain—“Glymphatic” Pathway Clears Aβ, Requires Water Channel
Biomarker Meta Analysis Citations
Paper Citations
- Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013 Feb;12(2):207-16. PubMed.
- Chiasserini D, Parnetti L, Andreasson U, Zetterberg H, Giannandrea D, Calabresi P, Blennow K. CSF levels of heart fatty acid binding protein are altered during early phases of Alzheimer's disease. J Alzheimers Dis. 2010;22(4):1281-8. PubMed.
- Guo LH, Alexopoulos P, Perneczky R. Heart-type fatty acid binding protein and vascular endothelial growth factor: cerebrospinal fluid biomarker candidates for Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci. 2013 Oct;263(7):553-60. Epub 2013 Apr 17 PubMed.
- Ghani F, Wu AH, Graff L, Petry C, Armstrong G, Prigent F, Brown M. Role of heart-type fatty acid-binding protein in early detection of acute myocardial infarction. Clin Chem. 2000 May;46(5):718-9. PubMed.
- Toledo JB, Toledo E, Weiner MW, Jack CR, Jagust W, Lee VM, Shaw LM, Trojanowski JQ, . Cardiovascular risk factors, cortisol, and amyloid-β deposition in Alzheimer's Disease Neuroimaging Initiative. Alzheimers Dement. 2012 Nov;8(6):483-9. PubMed.
- Erqou S, Thompson A, Di Angelantonio E, Saleheen D, Kaptoge S, Marcovina S, Danesh J. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010 May 11;55(19):2160-7. PubMed.
- Oxenkrug G. Interferon-gamma - Inducible Inflammation: Contribution to Aging and Aging-Associated Psychiatric Disorders. Aging Dis. 2011 Dec;2(6):474-86. Epub 2011 Dec 2 PubMed.
- Bodnar RJ, Yates CC, Wells A. IP-10 blocks vascular endothelial growth factor-induced endothelial cell motility and tube formation via inhibition of calpain. Circ Res. 2006 Mar 17;98(5):617-25. Epub 2006 Feb 16 PubMed.
- Li Y, Zhou C, Zhou X, Li L, Hui R. Pregnancy-associated plasma protein A predicts adverse vascular events in patients with coronary heart disease: a systematic review and meta-analysis. Arch Med Sci. 2013 Jun 20;9(3):389-97. Epub 2013 May 28 PubMed.
- Weller RO, Hawkes CA, Carare RO, Hardy J. Does the difference between PART and Alzheimer's disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Aβ and propagation of tau?. Acta Neuropathol. 2015 May;129(5):763-6. Epub 2015 Mar 27 PubMed.
- Bakker EN, Bacskai BJ, Arbel-Ornath M, Aldea R, Bedussi B, Morris AW, Weller RO, Carare RO. Lymphatic Clearance of the Brain: Perivascular, Paravascular and Significance for Neurodegenerative Diseases. Cell Mol Neurobiol. 2016 Mar;36(2):181-94. Epub 2016 Mar 18 PubMed.
- Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao Y, Deane R, Nedergaard M. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci. 2013 Nov 13;33(46):18190-9. PubMed.
External Citations
Further Reading
Papers
- Young AL, Oxtoby NP, Daga P, Cash DM, Fox NC, Ourselin S, Schott JM, Alexander DC, Alzheimer’s Disease Neuroimaging Initiative. A data-driven model of biomarker changes in sporadic Alzheimer's disease. Brain. 2014 Sep;137(Pt 9):2564-77. Epub 2014 Jul 9 PubMed.
- Roher AE. Cardiovascular system participation in Alzheimer's disease pathogenesis. J Intern Med. 2014 Sep 22; PubMed.
- Choi HJ, Seo EH, Yi D, Sohn BK, Choe YM, Byun MS, Lee JM, Woo JI, Lee DY. Amyloid-Independent Amnestic Mild Cognitive Impairment and Serum Apolipoprotein A1 Levels. Am J Geriatr Psychiatry. 2015 Jun 26; PubMed.
Primary Papers
- Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. 2016 Jun 21;7:11934. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Washington University
I’m not an expert on the mathematical modeling done here. While it’s clear that vascular abnormalities can contribute to cognitive decline, I am very concerned about the conclusion drawn from this data, namely that vascular dysregulation is the earliest event leading to LOAD.
One concern is that if you look at the results of their data analysis in Figure 3, it shows that Aβ42 changes after amyloid deposition detected by amyloid imaging using florbetapir. Florbetapir is a good amyloid imaging agent but it is not sensitive. We know that CSF Aβ42 begins to drop right at or likely before amyloid imaging positivity as assessed in longitudinal and cross-sectional studies done with PiB (a more sensitive amyloid imaging agent than florbetapir). It is also clear from several longitudinal and cross-sectional studies that functional decline does not come before decreases in CSF Aβ42 (shown in Figure 3). I am concerned that either the modeling used or the way the analysis from the ADNI dataset was done has led to the wrong conclusions. The ADNI dataset has more individuals who are impaired than non-impaired. Also, the subjects are relatively old at entry. It would be better to try to model this type of data from longitudinal studies that start with people at younger ages (40-70), when AD pathology is usually beginning, and who develop clinical disease in their 70s and 80s. This is being done in a number of data sets.
Weill Cornell Medical College
Iturria-Medina and colleagues report a new analysis of the ADNI data that intriguingly suggests an early role for vascular dysregulation in AD pathophysiology. Their approach is a tour de force in the field of progression tracking. Although others have used similar techniques for aligning biomarkers along a common age axis, this study stands out in its thoroughness and in the amount of data sets and biomarkers used.
That vascular effects precede other abnormalities is easily the most interesting finding. This idea has of course been proposed before and this study provides a strong statistical support to it. Practically, these results might persuade investigators to propose the inclusion of high quality MR perfusion sequences in future research and clinical protocols.
To me what is even more interesting is that this study allows new mathematical models of progression to be encoded and tested. For instance, vascular maps could be used as starting patterns that could drive models of subsequent brain-wide spread using the Network Diffusion framework or other models of networked spread.
NYU School of Medicine
The paper by Iturria-Medina et al. reminds us how little we know about the biological predictors, modulators, correlates, and causes contributing to late-onset Alzheimer type dementia (LOAD). Their multimodality study based on the entire ADNI cohort reveals that among the multiple domains of acquired data, vascular factors carry the greatest weight in characterizing the trajectory of cognitive decline. Moreover, these data challenge existing views of the primacy of Aβ lesions as the first detectable feature of the LOAD process. Rather, the authors point us toward vascular-mediated defects that have a role in the clearance of brain waste products, which of course includes products of Aβ metabolism. This paper should encourage us to consider the subject composition from large-scale national projects, the biological measures to invest in, and the evolution of high dimensional data analytic procedures such as first presented here.
University of Southern California
University of Southern California
Iturria-Medina and colleagues provide definitive confirmation of two facts about the Alzheimer's pathophysiological cascade that have been underappreciated, despite cumulative evidence from several groups: First, that memory dysfunction may occur prior to abnormalities in biomarkers for amyloid and tau, the classic pathophysiological hallmarks of Alzheimer's disease. This suggests that another, earlier pathology may initiate cognitive decline. This leads us to their second finding, which is that changes in the cerebrovascular system (vascular dysregulation) precede cerebral amyloidosis and tau-mediated neurodegeneration, providing a potential explanation for early cognitive changes. Their findings concur very well with our recent observations that loss of cerebrovascular integrity and blood-brain barrier breakdown are amongst the first changes in the brain during normal aging and in individuals with mild cognitive impairment (Montagne et al., 2015). Given that findings by Iturria-Medina et al. are from one of the largest and most comprehensive biomarker analyses ever conducted, it is probably good time for us to begin thinking how to update our model of the Alzheimer's pathophysiological cascade by including vascular dysfunction as a driver of disease pathogenesis.
References:
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015 Jan 21;85(2):296-302. PubMed.
University of Southampton School of Medicine
In the paper Iturria-Medina et al. analyzed 7,700 multimodality brain images and tens of different plasma and CSF biomarkers from 1,171 healthy patients and from patients with late-onset Alzheimer's disease (LOAD). A data-driven approach revealed multiple factors responsible for the progression of Alzheimer's disease. One of the major conclusions is that vascular dysregulation may be the earliest and strongest pathological factor associated with LOAD, followed in order by deposition of Aβ, dysregulation of glucose metabolism, functional impairment, and atrophy of the gray matter
Such an approach promises to be extremely valuable, particularly as it is difficult to determine the causative factors in Alzheimer's disease from those factors that are secondary effects. The data-driven approach using an evaluation of misfolded amyloid proteins, glucose metabolism, cerebral blood flow, functional activity, and neuroimaging revealed a characteristic trajectory for each biological factor during the development of Alzheimer's disease.
Valuable data have been gathered from the pathological examination of the brains, CSF, and blood over the last 100 years. The present data-driven approach is a valuable addition for those seeking to determine the major factors that initiate Alzheimer's disease and result in its progression. The hypothesis that vascular dysregulation may be the earliest and strongest pathological factor in Alzheimer's disease requires further investigation. Arteries and capillaries in the brain not only deliver blood and nutrients, they are also the route for the elimination of fluid and soluble metabolites from the brain in the maintenance of tissue homoeostasis. Although the authors of the paper recognized that failure of elimination of Aβ is a major factor in the pathogenesis of Alzheimer's disease, they concentrate on absorption of Aβ into the blood. In assessing the role of arteries and capillaries in the brain, the perivascular route for elimination of fluid and solutes, including Aβ, should not be ignored. Experimental studies have shown that perivascular elimination of fluid and solutes is impaired with increasing age, and this is reflected in the accumulation of Aβ in the walls of capillaries and arteries as cerebral amyloid angiopathy in Alzheimer's disease (Carare et al., 2013; Weller et al., 2015).
The current paper is a significant step forward in our understanding of the pathogenesis and progression of Alzheimer's disease. Its value will be increased by correlating the data with previous observations, particularly on the pathophysiology of cerebral arteries and capillaries in the elimination of soluble metabolites, including Aβ in the maintenance of homoeostasis of the brain.
References:
Carare RO, Hawkes CA, Jeffrey M, Kalaria RN, Weller RO. Cerebral amyloid angiopathy, Prion angiopathy, CADASIL and the spectrum of Protein Elimination-Failure Angiopathies (PEFA) in neurodegenerative disease with a focus on therapy. Neuropathol Appl Neurobiol. 2013 Mar 13; PubMed.
Weller RO, Hawkes CA, Carare RO, Hardy J. Does the difference between PART and Alzheimer's disease lie in the age-related changes in cerebral arteries that trigger the accumulation of Aβ and propagation of tau?. Acta Neuropathol. 2015 May;129(5):763-6. Epub 2015 Mar 27 PubMed.
Maastricht University; VU University Medical Centre
One remarkable finding of this interesting study is the observation that the CSF Aβ1-42 abnormality index only increases to 30 percent at the late AD stage. However, amyloid PET binding (which correlates around 0.8 with CSF Aβ1-42 in ADNI [Toledo et al., 2015]) reaches a much higher abnormality index than CSF Aβ1-42, namely around 90 percent. One possible explanation could be that CSF Aβ1-42 plateaus earlier than amyloid PET. As pointed out above, a more detailed definition of the abnormality index would be useful to better understand these findings.
References:
Toledo JB, Bjerke M, Da X, Landau SM, Foster NL, Jagust W, Jack C Jr, Weiner M, Davatzikos C, Shaw LM, Trojanowski JQ, Alzheimer’s Disease Neuroimaging Initiative Investigators. Nonlinear Association Between Cerebrospinal Fluid and Florbetapir F-18 β-Amyloid Measures Across the Spectrum of Alzheimer Disease. JAMA Neurol. 2015 May;72(5):571-81. PubMed.
RIKEN Center for Brain Science
I tend to agree with David Holtzman. One thing unclear to me is why the authors did not distinguish between ApoE4 carriers and non-carriers in their mathematical modeling. This is because ApoE4 genotype is an independent risk factor for vascular dysfunction and also for CAA. The errors that arose during computation might have been smaller and could have given different conclusions.
Saba University School of Medicine
This study is very impressive. The list of consortium members is gigantic, and the amount of work amazing. There are, however, a couple of points that trigger me to comment on such an impressive piece of work and the role it assigns to early vascular dysregulation on LOAD.
The authors themselves are very careful and warn not to over-interpret the results. They state this clearly: “Although the obtained abnormality trajectories may be reflecting a tentative ordering in which pathophysiological events occur, our results should be interpreted more in terms of biomarker sensitivity to disease progression than in terms of causal pathologic interactions conducive of LOAD.”
Nevertheless, you immediately read in a comment on this paper that “memory dysfunction may occur prior to abnormalities in biomarkers for amyloid and tau”—something that is in direct contradiction to many other findings, as pointed out by another commenter. Many readers of this paper will make the same kind of mistake to find support in this large study for their alternative hypotheses to explain LOAD.
However, detecting a fever before the causative pathogen can been detected does not mean that the body temperature increases before the infection. It just means that you can measure the increase in body temperature before you can detect the causative infectious agent, but the infection always occurs first.
Many scientists just want to believe that AD is a highly complex disorder that must have a high number of complex mechanisms underlying the disease. Although I do not reject this hypothesis and I am convinced that to some extent it is true, it is always interesting to remember that AD is one of the few common diseases that can be explained (in some familial cases) by the simplest mechanism known in medicine. One single base substitution in a single allele of one single gene (that is, a single base substitution in the 6 billion bases of a diploid genome) can be sufficient to cause an autosomal-dominant form of AD that is basically indistinguishable from the more common LOAD.
This is, of course, in the case of a mutation of the APP gene. We also know that an extra copy of the wild-type APP gene that leads to a 50 percent increase in APP production without any other alteration can also cause a form of AD that is indistinguishable from LOAD. Actually, we do not know many common complex disorders that can be explained by such a minimal, simple, and straightforward etiology.
The big problem with AD is that, although in this familiar form of the disease everything (and I mean really everything) about the disease can be explained by just an overexpression of APP by 50 percent—the plaques, tangles, neuronal cell death, cognitive decline, etc., etc., etc.—it seems amazing that when you ask what could be causing the overproduction of APP in LOAD, a convincing answer is nowhere to be found. On the contrary, the authors of this impressive paper state that even if their approach “does not reveal causal pathologic interactions, concordant evidence suggests that in LOAD Aβ deposition is mainly caused by a deficiency in the Aβ clearance system rather than by an Aβ overproduction.” They probably favor such an explanation to justify the important role they assign to the early vascular dysregulation described in their impressive piece of work.
There is, however, nothing concordant about Aβ deposition being caused mainly by a deficiency in clearance rather that production. As indicated above, everything about this disease can be explained by a simple overproduction of APP and thus also the observed decrease in clearance. The authors themselves indicate that “Aβ has vascular destructive activity”.
The reason so many studies overlook the increase in APP production is because they simply forget to take into account that, in AD brains, an apparently similar level of APP is produced (in some regions) by less than half the number of neurons. On a per-neuron basis, the production may well have doubled. Note that even if this is overlooked, not taken into account, or not detected by a majority of studies, the logic is indisputable. If in AD brains fewer neurons produce the same amount of APP, then the production at the individual neuron level has to increase. It seems reasonable to assume that overproduction at the individual neuron level is toxic to the neuron itself or its direct neighbors.
In my opinion the AD field too often focuses on the consequences of the disease instead of the causes. I have to admit, from analyzing a plane wreck after a crash it would be difficult to deduce what caused the accident. If the black box indicates a leak in the fuel tank, however, you have a strong clue. The experiments of nature that clearly provide us clues as to what has to happen first and what should follow in AD, irrespective of the sensitivity of the assay used, should be more consistently used in the interpretation of complex experimental results if we are going to find a cure soon for this devastating disorder. There is not much time to lose, as we are unfortunately not getting any younger.
Johns Hopkins University
This is an interesting report, and should be considered in the context of a similar observation that was recently published on FAD subjects (Lee et al., 2016), in which white matter hyperintensities were observed as more prominent in FAD mutation carriers within the DIAN cohort than in related non-carriers, increasing approximately six years before expected symptom onset, with divergence from non-mutation-carrying family members as much as 22 years before symptom onset.
The findings in that publication were interpreted as suggesting that WMHs are a core feature of AD that should be integrated into pathogenic models of the disease, as well as a potential therapeutic target. Together, these reports speak to vascular abnormalities as contributing to early disease pathogenesis, perhaps as early as the onset of amyloid accumulation. An alternative view might be that amyloid is still driving the disease in the FAD mutation carriers, but with an early prodromal outcome being induction of vascular lesions.
References:
Lee S, Viqar F, Zimmerman ME, Narkhede A, Tosto G, Benzinger TL, Marcus DS, Fagan AM, Goate A, Fox NC, Cairns NJ, Holtzman DM, Buckles V, Ghetti B, McDade E, Martins RN, Saykin AJ, Masters CL, Ringman JM, Ryan NS, Förster S, Laske C, Schofield PR, Sperling RA, Salloway S, Correia S, Jack C Jr, Weiner M, Bateman RJ, Morris JC, Mayeux R, Brickman AM, Dominantly Inherited Alzheimer Network. White matter hyperintensities are a core feature of Alzheimer's disease: Evidence from the dominantly inherited Alzheimer network. Ann Neurol. 2016 Jun;79(6):929-39. Epub 2016 Apr 27 PubMed.
University of Minnesota, Twin Cities
I read the paper by Iturria-Medina et al. and the discussions here with great interest, as the research in my laboratory has been focused on the connections between cardiovascular disease and Alzheimer’s disease. While most of the molecules/biomarkers described are clear, there may be some confusion about apo(a) and apoA-I. Please note that these are two different proteins; they are encoded by different genes and are different in structure and function (McLean et al., 1987; Segrest et al., 2000). To avoid any potential confusions, apo(a) has been called the “apo little a” as written in the cardiovascular field. However, apo(a) is called Apo A in the paper by Iturria-Medina et al. Apo(a) associates with low-density lipoproteins (LDL), not high-density lipoproteins (HDL) as described by Iturria-Medina and colleagues. Apo(a) forms lipoprotein particles called lipoprotein(a) [Lp(a)]. ApoA-I is the major protein component of HDLs in the plasma and determines most of HDL functions.
It has been well established that high levels of apoA-I/HDL reduce the risk of cardiovascular disease, whereas high levels of apo(a)/Lp(a) increase the risk of cardiovascular disease (Davidson and Toth, 2007; Emerging Risk Factors et al., 2009). The cardiovascular risk associated with apo(a)/Lp(a) is further complicated by the size variations of apo(a), due to the existence of polymorphic repeats in its gene (Erqou et al., 2010). In the paper by Iturria-Medina et al., it was the level of apo(a) in the CSF that was associated with the risk of Alzheimer’s disease.
Previously, other clinical studies have shown that high levels of plasma apoA-I/HDL are associated with better cognitive function and a reduced risk of Alzheimer’s disease (reviewed in Hottman et al., 2014). We and others have demonstrated that genetic manipulation of apoA-I affects the extent of cerebral amyloid angiopathy (CAA) and neuroinflammation in animal models of Alzheimer’s disease (Lefterov et al., 2010; Lewis et al., 2010; Oct 2010 news). However, few studies have investigated the role of apo(a)/Lp(a) in cognition and AD. Since apo(a) and apoA-I play opposite roles in cardiovascular disease and most likely also in Alzheimer’s disease, we should avoid any confusions when these two proteins are discussed.
References:
Davidson MH, Toth PP. High-density lipoprotein metabolism: potential therapeutic targets. Am J Cardiol. 2007 Dec 3;100(11 A):n32-40. PubMed.
Emerging Risk Factors Collaboration, Erqou S, Kaptoge S, Perry PL, Di Angelantonio E, Thompson A, White IR, Marcovina SM, Collins R, Thompson SG, Danesh J. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009 Jul 22;302(4):412-23. PubMed.
Erqou S, Thompson A, Di Angelantonio E, Saleheen D, Kaptoge S, Marcovina S, Danesh J. Apolipoprotein(a) isoforms and the risk of vascular disease: systematic review of 40 studies involving 58,000 participants. J Am Coll Cardiol. 2010 May 11;55(19):2160-7. PubMed.
Hottman DA, Chernick D, Cheng S, Wang Z, Li L. HDL and cognition in neurodegenerative disorders. Neurobiol Dis. 2014 Dec;72 Pt A:22-36. Epub 2014 Aug 13 PubMed.
Lefterov I, Fitz NF, Cronican AA, Fogg A, Lefterov P, Kodali R, Wetzel R, Koldamova R. Apolipoprotein A-I deficiency increases cerebral amyloid angiopathy and cognitive deficits in APP/PS1DeltaE9 mice. J Biol Chem. 2010 Nov 19;285(47):36945-57. Epub 2010 Aug 25 PubMed.
Lewis TL, Cao D, Lu H, Mans RA, Su YR, Jungbauer L, Linton MF, Fazio S, LaDu MJ, Li L. Overexpression of human apolipoprotein A-I preserves cognitive function and attenuates neuroinflammation and cerebral amyloid angiopathy in a mouse model of Alzheimer disease. J Biol Chem. 2010 Nov 19;285(47):36958-68. Epub 2010 Sep 16 PubMed.
McLean JW, Tomlinson JE, Kuang WJ, Eaton DL, Chen EY, Fless GM, Scanu AM, Lawn RM. cDNA sequence of human apolipoprotein(a) is homologous to plasminogen. Nature. 1987 Nov 12-18;330(6144):132-7. PubMed.
Segrest JP, Li L, Anantharamaiah GM, Harvey SC, Liadaki KN, Zannis V. Structure and function of apolipoprotein A-I and high-density lipoprotein. Curr Opin Lipidol. 2000 Apr;11(2):105-15. PubMed.
McGill University
A brief response to the interesting comments by Torik Ayoubi: An earlier paper by our group (Iturria-Medina et al., 2014) looked at a causal model of Aβ propagation through the white-matter pathways. The results suggest that accumulation of Aβ is more a consequence of reduced clearance than overproduction. Moreover, that clearance deficit showed a strong gene-dose dependence upon the number of APOE4 alleles.
References:
Iturria-Medina Y, Sotero RC, Toussaint PJ, Evans AC, Alzheimer's Disease Neuroimaging Initiative. Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Comput Biol. 2014 Nov;10(11):e1003956. Epub 2014 Nov 20 PubMed.
Make a Comment
To make a comment you must login or register.