Huntingtin Repeats Cause—FTD, Caveolin Variants—ALS?
Quick Links
Geneticists have added two more genes to the list of genetic risk factors for amyotrophic lateral sclerosis and frontotemporal dementia. In Cell Reports on December 1, Johnathan Cooper-Knock, Pamela Shaw, and colleagues at the University of Sheffield, U.K., homed in on enhancer elements, searching for mutations that regulate gene expression. Mutations in the enhancer region controlling caveolin 1 expression popped up in about 1 percent of ALS cases. These mutations reduced CAV1 expression in patient cells, resulting in altered cell-membrane lipids and disrupted cell signaling. The researchers also found point mutations that changed some of the protein’s amino acids associated with ALS. Together, the enhancer and coding regions variants flag CAV1 as an ALS risk gene.
- Polyglutamine-expanded huntingtin inclusions found postmortem in ALS/FTD tissue.
- CAV1 enhancer mutations disrupt membrane lipids and cell signaling in human cells.
- Both types of mutation are rare but could have clinical implications.
In Neuron on November 25, researchers in Bryan Traynor’s lab at the National Institute on Aging, Bethesda, Maryland, reported finding trinucleotide expansions in the huntingtin gene in people who have ALS or FTD. Also analyzing whole-genome sequence data, they found that about 0.1 percent of cases had polyglutamine repeats in huntingtin long enough to cause Huntington’s disease. These eight carriers had shown no classic HD symptoms or atrophy of the striatum that typifies the disease. Instead, postmortem analysis revealed TDP-43 aggregates in the frontal cortex, which is typical for ALS/FTD, but also huntingtin aggregates there.
Since drugs targeting CAV1 and polyglutamine-expanded HTT are being tested in clinical trials, screening people with ALS and FTD for those mutations may expand treatment options, researchers suggested.
“Noncoding CAV1 variants contributing to ALS is not surprising, but the possible link between HTT and ALS-FTD is intriguing, given the clear links to other expansions in ATXN2 and C9ORF72,” Jemeen Sreedharan, King’s College London, told Alzforum (see full comment below).
Only 10 percent of sporadic ALS cases have been associated with a genetic mutation (Chia et al., 2018). In search of more genetic ties, Cooper-Knock and colleagues looked beyond coding regions. Could variants there have implications in ALS?
To find out, the researchers searched whole-genome sequencing (WGS) data for enhancer variants in CAV1, which encodes caveolin 1, a neurotrophic signaling and survival protein (Head et al., 2011). In fact, overexpressing caveolin 1 in an ALS mouse model reduced motor neuron death and improved survival, and researchers are pursuing this approach as a potential therapy (Sawada et al., 2019).
Cooper-Knock and colleagues found more than 150 point mutations at the CAV1/CAV2 loci in 56 of 4,495 ALS cases that lay in prospective active enhancer regions of a variety of cell types. In contrast, only two of 1,925 controls carried such genetic changes. When the researchers looked at neuronal enhancers, specifically, CAV1/CAV2 variants were associated with the disease, appearing in five times as many people with ALS than in controls. Twelve CAV1 coding mutations also occurred in 15 ALS cases but only one control. The researchers believe they may impair caveolin 1 function.
To find out if the enhancer variants altered function, the scientists examined white blood cells from two of those 56 ALS cases. Cells harboring the variant chr7:116222625T>C made less caveolin 1 than did cells from three healthy controls, cells from three people with ALS who did not have this mutation, and cells harboring a different enhancer variant. Cells with the 116222625T>C variant, and those from ALS cases carrying CAV1 coding mutations, also had fewer GM1 ganglioside membrane lipids (see image below). GM1 gangliosides are neurotrophic, supporting neuron function from their location within lipid rafts (Chiricozzi et al., 2020). Caveolin 1 helps cells maintain these membrane rafts, which are crucial for cell signaling (Sawada et al., 2019). In fact, ganglioside level correlated with caveolin 1 expression together in all the white blood cell lines tested, suggesting these lipid rafts depend on the protein.
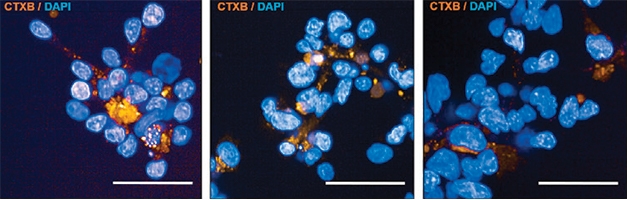
Lipidless. Lymphoblastoid cells derived from a healthy control (left) or a person with ALS who did not carry the CAV1 enhancer variant, 116222625T>C (middle), take up cholera toxin B (gold), which integrates into membrane lipid rafts. Cells from a person with ALS (right) who carried the CAV1 variant bound less of the toxin. [Courtesy of Cooper-Knock et al., Cell Reports, 2020.]
What about in neurons? Cooper-Knock used CRISPR/Cas9 to introduce insertions or deletions near the ALS variants in the CAV1 enhancer in SH-SY5Y cells. A single nucleotide insertion docked expression of caveolin 1 by 500-fold, showing that mutations at this spot can modulate gene expression in neurons. In silico analysis suggested this region binds five transcription factors: RAD21, CTCF, FOS, SMC3, and CEBPB.
Cooper-Knock claimed this is the first large-scale association between noncoding mutations and ALS. “Variants in untranslated regions and promoters that influence expression of ALS-linked genes will be increasingly found to play a role in ALS-FTD,” he predicted.
In the second study, first author Ramita Dewan and colleagues in Traynor’s lab asked if gene expansions might associate with ALS and FTD. Many diseases have been linked to such expansions, including the hexanucleotide repeats found in intron one of C9ORF72 in people with ALS/FTD, and the trinucleotide repeats in the huntingtin gene (HTT) in that eponymous disease (Majounie et al., 2012; MacDonald et al., 1993). Indeed, expansions in ataxin 2, which cause a form of ataxia, are also a risk factor for ALS/FTD (Wang et al., 2016).
Dewan and colleagues studied whole-genome sequencing data from 1,065 people with ALS, 1,377 with FTD, 2,599 with Lewy body dementia, and 3,158 older controls. They searched for expansions in 50 genes associated with neurodegenerative diseases, including C9ORF72, ATXN2, and HTT. As expected, the ALS and FTD samples turned up repeats in C9ORF72, as this is the most common genetic cause of both (Majounie et al., 2012).
The researchers found but one other expansion. Much to their surprise, it was the trinucleotide CAG repeats that cause HD. They appeared in three of the 2,442 ALS/FTD cases (see image below). At 40 to 41 repeats, the expansions were on the lower end of the pathogenic range. People with more the 36 repeats are at high risk for Huntington’s, while those with more than 40 are virtually guaranteed to develop the disease (Chong et al., 1997). Aside from the C9ORF72 mutation, Dewan and colleagues found no other expansions in these three cases.

HTT Repeats. Pathogenic huntingtin’s expansions, namely those longer than 40 repeats (orange), cropped up in people with ALS or FTD , but not in people with Lewy body disease, or healthy controls. [Courtesy of Dewan et al., Neuron, 2020.]
Huntingtin being involved in ALS and FTD comes as a surprise. Expanded HTT typically causes neurodegeneration in the striatum, which is largely spared in ALS/FTD, and previous work found no such link (Ramos et al., 2012). Interestingly, striatal atrophy had previously been seen in people with the behavioral variant of FTD (Prado et al., 2015).
To be sure of this association, Dewan and colleagues searched WGS data from another 2,665 people with ALS, 1,009 with FTD, and 210 age-matched controls. Again, they found HTT repeats larger than 40 in five cases.
Of the eight people with HTT expansions, five had FTD, three ALS. One was a woman who had been diagnosed with FTD when she was 17 but got a young-onset HD diagnosis a few years later when a genetic test uncovered 64 HTT repeats. Her father, who suffered cognitive decline, a gait disorder, and motor speech disorder in his late 20s, was diagnosed with Huntington’s after his daughter’s genetic diagnosis.
None of the other seven developed movement disorders typical of HD or had a family history of that disease, although three did have family histories of neurological disorders. “It would be interesting to sequence the relatives of these ALS and FTD cases,” Sreedharan told Alzforum.
The researchers compared the frequency of HTT expansions to published data from five European population-based cohorts and the UK 100K Genomes Project (Gardiner et al., 2019; Peplow, 2016). Only 10 out of 31,463 people in the general population had 40 or more HTT repeats, meaning people with ALS or FTD are 4.4 times more likely to carry them.
“These findings are interesting but need to be confirmed in a large number of cases and controls,” Cooper-Knock told Alzforum. “It is important to remember that association does not prove causation.” Sreedharan agreed: “It remains unclear if HTT expansions are mechanistically involved in disease causation.”
To get a look at what was happening in the brain, Dewan and colleagues studied postmortem brain tissue from two of the ALS cases with HTT expansions: a woman and man who developed symptoms at age 56 and 61, respectively. Both had mild atrophy of precentral gyri, spinal motor neuron loss, and TDP-43 inclusions in their prefrontal cortices, all signs of ALS. However, they also had polyglutamine inclusions and huntingtin aggregates in their prefrontal cortices, which is common in HD, but not in ALS or FTD. They had no atrophy, neuron loss, or active gliosis (see image below) in the striatum.
The researchers also examined the prefrontal cortices of three people with ALS/FTD who did not have HTT expansions, one person with diffuse Lewy body dementia, two with AD, and four healthy controls—none had huntingtin aggregates.

Mixed Pathology. In the brain of a person who carried HTT repeats and had ALS, TDP-43 inclusions (black arrows, left), huntingtin aggregates (black arrows, center), and ubiquitin aggregates (red arrows, center) were apparent in the prefrontal cortex. Polyglutamine inclusions were also found in the striatum (black arrows, right), but no atrophy. [Courtesy of Dewan et al., Neuron, 2020.]
“It would be interesting to investigate if there is a mechanistic link between TDP-43 and HD,” Sreedharan added. He is also curious if HTT repeats would be found in people with Alzheimer’s disease. Researchers previously identified a prion-like domain in TDP-43 that binds polyglutamine (Jul 2010 news).
Could the ALS and FTD cases with pathogenic HTT repeats have had atypical HD and been misdiagnosed? Their intact striatum makes this unlikely. What about them having both FTD or ALS and HD? The prevalence of all three diseases is low, at 22, six, and three per 100,000 people, respectively. According to the authors, there would only be three cases of co-occurrence in the entire U.S. population, whereas 10 people have been documented before to have both ALS and HD (Chhetri et al., 2014). However, HTT repeats had not been found previously in people with only ALS or FTD.
“These findings have massive implications, especially for screening for HTT repeats in people with ALS or FTD and counseling them and their families,” Cooper-Knock said. That is because these rare ALS/FTD cases might benefit from HTT antisense oligonucleotide (ASO) therapies currently progressing through clinical trials (Tabrizi et al., 2020). Wave Life Sciences has two allele-specific ASOs, WVE-120101 and WVE-120102 in Phase 1/2 (NCT03225833 and NCT03225846). Roche has Tominersen in Phase 3 (NCT03761849).—Chelsea Weidman Burke
References
News Citations
Paper Citations
- Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: diagnostic and clinical implications. Lancet Neurol. 2018 Jan;17(1):94-102. Epub 2017 Nov 16 PubMed.
- Head BP, Hu Y, Finley JC, Saldana MD, Bonds JA, Miyanohara A, Niesman IR, Ali SS, Murray F, Insel PA, Roth DM, Patel HH, Patel PM. Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J Biol Chem. 2011 Sep 23;286(38):33310-21. PubMed.
- Sawada A, Wang S, Jian M, Leem J, Wackerbarth J, Egawa J, Schilling JM, Platoshyn O, Zemljic-Harpf A, Roth DM, Patel HH, Patel PM, Marsala M, Head BP. Neuron-targeted caveolin-1 improves neuromuscular function and extends survival in SOD1G93A mice. FASEB J. 2019 Jun;33(6):7545-7554. Epub 2019 Mar 20 PubMed.
- Chiricozzi E, Lunghi G, Di Biase E, Fazzari M, Sonnino S, Mauri L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int J Mol Sci. 2020 Jan 29;21(3) PubMed.
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chiò A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, , Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S, Traynor BJ. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012 Apr;11(4):323-30. PubMed.
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993 Mar 26;72(6):971-83. PubMed.
- Wang MD, Little J, Gomes J, Cashman NR, Krewski D. Identification of risk factors associated with onset and progression of amyotrophic lateral sclerosis using systematic review and meta-analysis. Neurotoxicology. 2016 Jul 1; PubMed.
- Chong SS, Almqvist E, Telenius H, LaTray L, Nichol K, Bourdelat-Parks B, Goldberg YP, Haddad BR, Richards F, Sillence D, Greenberg CR, Ives E, Van den Engh G, Hughes MR, Hayden MR. Contribution of DNA sequence and CAG size to mutation frequencies of intermediate alleles for Huntington disease: evidence from single sperm analyses. Hum Mol Genet. 1997 Feb;6(2):301-9. PubMed.
- Ramos EM, Keagle P, Gillis T, Lowe P, Mysore JS, Leclerc AL, Ratti A, Ticozzi N, Gellera C, Gusella JF, Silani V, Alonso I, Brown RH Jr, MacDonald ME, Landers JE. Prevalence of Huntington's disease gene CAG repeat alleles in sporadic amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler. 2012 May;13(3):265-9. Epub 2012 Mar 13 PubMed.
- Prado LG, Bicalho IC, Magalhães D, Caramelli P, Teixeira AL, de Souza LC. C9ORF72 and the FTD-ALS spectrum: A systematic review of neuroimaging studies. Dement Neuropsychol. 2015 Oct-Dec;9(4):413-421. PubMed.
- Gardiner SL, Boogaard MW, Trompet S, de Mutsert R, Rosendaal FR, Gussekloo J, Jukema JW, Roos RA, Aziz NA. Prevalence of Carriers of Intermediate and Pathological Polyglutamine Disease-Associated Alleles Among Large Population-Based Cohorts. JAMA Neurol. 2019 Jun 1;76(6):650-656. PubMed.
- Peplow M. The 100,000 Genomes Project. BMJ. 2016 Apr 13;353:i1757. PubMed.
- Chhetri SK, Dayanandan R, Bindman D, Craufurd D, Majeed T. Amyotrophic lateral sclerosis and Huntington's disease: neurodegenerative link or coincidence?. Amyotroph Lateral Scler Frontotemporal Degener. 2014 Mar;15(1-2):145-7. Epub 2013 Jul 16 PubMed.
- Tabrizi SJ, Flower MD, Ross CA, Wild EJ. Huntington disease: new insights into molecular pathogenesis and therapeutic opportunities. Nat Rev Neurol. 2020 Oct;16(10):529-546. Epub 2020 Aug 14 PubMed.
External Citations
Further Reading
Papers
- Menéndez-González M, Clarimón J, Rosas-Allende I, Blázquez M, San Martín ES, García-Fernández C, Lleó A, Dols-Icardo O, Illán-Gala I, Morís G, Ribacoba R, Álvarez V, Martínez C. HTT gene intermediate alleles in neurodegeneration: evidence for association with Alzheimer's disease. Neurobiol Aging. 2019 Apr;76:215.e9-215.e14. Epub 2018 Nov 28 PubMed.
- Rosas I, Martínez C, Clarimón J, Lleó A, Illán-Gala I, Dols-Icardo O, Borroni B, Almeida MR, van der Zee J, Van Broeckhoven C, Bruni AC, Anfossi M, Bernardi L, Maletta R, Serpente M, Galimberti D, Scarpini E, Rossi G, Caroppo P, Benussi L, Ghidoni R, Binetti G, Nacmias B, Sorbi S, Piaceri I, Bagnoli S, Antonell A, Sánchez-Valle R, De la Casa-Fages B, Grandas F, Diez-Fairen M, Pastor P, Ferrari R, Álvarez V, Menéndez-González M. Role for ATXN1, ATXN2, and HTT intermediate repeats in frontotemporal dementia and Alzheimer's disease. Neurobiol Aging. 2020 Mar;87:139.e1-139.e7. Epub 2019 Nov 1 PubMed.
Primary Papers
- Cooper-Knock J, Zhang S, Kenna KP, Moll T, Franklin JP, Allen S, Nezhad HG, Iacoangeli A, Yacovzada NY, Eitan C, Hornstein E, Elhaik E, Celadova P, Bose D, Farhan S, Fishilevich S, Lancet D, Morrison KE, Shaw CE, Al-Chalabi A, Project MinE ALS Sequencing Consortium, Veldink JH, Kirby J, Snyder MP, Shaw PJ. Rare Variant Burden Analysis within Enhancers Identifies CAV1 as an ALS Risk Gene. Cell Rep. 2020 Dec 1;33(9):108456. PubMed.
- Dewan R, Chia R, Ding J, Hickman RA, Stein TD, Abramzon Y, Ahmed S, Sabir MS, Portley MK, Tucci A, Ibáñez K, Shankaracharya FN, Keagle P, Rossi G, Caroppo P, Tagliavini F, Waldo ML, Johansson PM, Nilsson CF, American Genome Center (TAGC), FALS Sequencing Consortium, Genomics England Research Consortium, International ALS/FTD Genomics Consortium (iAFGC), International FTD Genetics Consortium (IFGC), International LBD Genomics Consortium (iLBDGC), NYGC ALS Consortium, PROSPECT Consortium, Rowe JB, Benussi L, Binetti G, Ghidoni R, Jabbari E, Viollet C, Glass JD, Singleton AB, Silani V, Ross OA, Ryten M, Torkamani A, Tanaka T, Ferrucci L, Resnick SM, Pickering-Brown S, Brady CB, Kowal N, Hardy JA, Van Deerlin V, Vonsattel JP, Harms MB, Morris HR, Ferrari R, Landers JE, Chiò A, Gibbs JR, Dalgard CL, Scholz SW, Traynor BJ. Pathogenic Huntingtin Repeat Expansions in Patients with Frontotemporal Dementia and Amyotrophic Lateral Sclerosis. Neuron. 2021 Feb 3;109(3):448-460.e4. Epub 2020 Nov 26 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
King's College London
On the CAV1 paper, I think this is very interesting work exploiting the power of WGS and the freedom of information/data offered through the ProjectMINE initiative, which is arguably the single most important advance in the ALS-FTD since the identification of C9ORF72 mutations.
That noncoding variants contribute to ALS-FTD is not surprising, but by focusing on enhancers they have isolated a credible gene in CAV1, which has already been linked to neurodegeneration. I think variants in UTRs and promoters that influence the expression level of other genes linked with ALS will be found to play a role in ALS-FTD.
On the HTT paper, the possibility of a link between HTT and ALS-FTD is intriguing given the clear links to other expansions in ATXN2 and C9ORF72. However, in this study, while they did find HTT expansions in patients, these were very rare, and the longest expansion was associated with a very atypical case, a 17-year-old who did not have FTD but actually the Westphal variant of HD.
Despite this, the patient continues to be included in their categorization as an FTD case in tables 1 and 2. The pathological data excluded huntingtin inclusions in the spinal cord but did intriguingly find inclusions in the prefrontal cortex. It was not clear how abundant these were and it remains unclear if HTT expansions are mechanistically involved in disease causation.
Nonetheless, it remains possible that cellular stresses invoked by an HTT expansion could contribute an additional “hit” to a cell that could act with other environmental/genetic factors and lead to ALS-FTD. This is further evidence of the value and the potential complexities that arise from WGS.
Make a Comment
To make a comment you must login or register.