How TDP-43’s Disheveled Tail Spells Trouble
Quick Links
Scientists at the National University of Singapore have published an atomic-level structure of the prion-like C-terminus of TDP-43, a protein associated with several neurodegenerative disorders. Their results confirm a highly disordered domain—similar to that of other RNA-binding proteins—that readily forms reversible oligomers. It is thought that this oligomerization helps organize RNA granules, which dynamically form and disperse in cells to control the translation of RNA.
Led by Jianxing Song, the researchers report that disease-related point mutations in this region of TDP-43 make the protein more likely to amass into amyloids or insoluble aggregates. Curiously, they identify a helical subdomain that interacts with membranes. They hypothesize that this association contributes to neurotoxicity and triggers amyotrophic lateral sclerosis (ALS). The results were published January 6 in PLoS Biology.
“It’s the first structural characterization of the whole prion-like domain of TDP-43, and the first study to look at the molecular effects of its disease-linked mutations,” said Nicolas Fawzi, Brown University, Providence, R.I., who was not involved in the research. He was curious whether the other 46 ALS-linked mutations identified in this region work by the same mechanism. He and colleagues had previously published the atomic structure of another prion-like domain of an RNA-binding protein, fused in sarcoma (FUS), and found a similarly disordered protein segment (Burke et al., 2015; Oct 2015 Webinar).
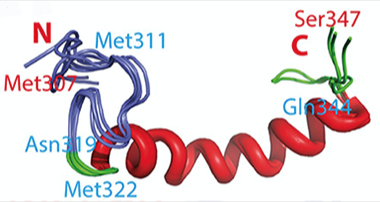
Membrane-Friendly:
Within the disordered C-terminus of TDP-43, a loop(purple) and an α-helix (red) interact with membranes. [Courtesy of Lim et al. 2015]
TDP-43 is a DNA- and RNA-binding protein that tends to misfold. Aggregates of the protein accumulate in neurons in patients with ALS, frontotemporal dementia, and Alzheimer’s disease. Its penchant for aggregation has complicated studies of its structure and physiological function. However, by using ultra-pure, salt-free water as a solvent, Song and colleagues kept this normally insoluble protein from binding to itself. He then resolved its native conformation. He had previously used the same strategy to solve the atomic-level structures of the ALS-linked proteins SOD1 and VAPB (Li et al., 2006; Lim et al., 2015; Qin et al, 2013). Most recently, he resolved the amino stub of TDP-43 (Dec 2014 news).
In the current study, first author Liangzhong Lim used a combination of circular dichroism (CD), fluorescence spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and electron miscopy to decode the minute details of the opposite end of the protein. The C-terminal region of TDP-43 is packed with the amino acids glutamine, asparagine, serine, and glycine, and has been blamed for the prion-like spread of ALS (for a review, see Polymenidou and Cleveland, 2011). This part of the protein also contains most of the known pathogenic mutations. To see how those polymorphisms affect this prion-like domain, the researchers compared the wild-type version with that of three ALS-linked mutants—A315E, Q331K, and M337V.
Lim and colleagues found the domain intrinsically disordered, lacking any fixed three-dimensional structure. It hangs loose and readily assembles into β-sheet-rich oligomers. While point mutations leave the overall structure mostly unchanged, they vary the peptide just enough so that it rapaciously assembles into amyloids. The results fit with studies reporting that mutations in the same regions of other DNA-binding proteins, such as FUS, make them more prone to aggregate (Murakami et al. 2015).
Previous reports suggest that prion-like domains in TDP-43 and FUS can bind nucleic acids (Han et al., 2012; Kato et al., 2012). Accordingly, Lim and colleagues found that single-stranded DNA enhanced oligomerization of wild-type TDP-43 and aggregation of the mutant forms.
Curiously, the authors found that in the presence of an artificial phospholipid bilayer, the area between residues Met311 and Gln343 folded into a membrane-burrowing structure consisting of an Ω-shaped loop and an α-helix. While several ALS-linked mutations lie in this region, none of those tested altered this structure. However, these mutants assembled into aggregates faster than did other TDP-43 variants. The authors suggest that the membrane-interacting subdomain helps concentrate the wild-type protein, favoring aggregation that may become toxic to cells.
“This interaction with the membrane is perhaps the most novel aspect of the work,” said Jim Shorter, University of Pennsylvania, Philadelphia. Song noted that other ALS-linked proteins, such as SOD1, acquire the ability to interact with the membrane and become toxic, and he suggested this might be a common mechanism for their cytotoxicity (Lim et al., 2014; Qin et al., 2013). However, Shorter and Fawzi noted that TDP-43 does not typically associate with the membrane and wondered if the interaction was physiological.
The results fit Shorter’s previous report that ALS-related mutations in the prion-like domain of TDP-43 promote aggregation in yeast (Johnson et al., 2009). If small molecules could make mutant forms of the protein less likely to aggregate, they may treat disorders such as ALS, Shorter speculated. He noted that Lim and colleagues looked only at an isolated portion of this protein; what happens in the context of full-length TDP-43 remains unclear.—Gwyneth Dickey Zakaib
References
Webinar Citations
News Citations
Paper Citations
- Burke KA, Janke AM, Rhine CL, Fawzi NL. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol Cell. 2015 Oct 15;60(2):231-41. Epub 2015 Oct 8 PubMed.
- Li M, Liu J, Ran X, Fang M, Shi J, Qin H, Goh JM, Song J. Resurrecting abandoned proteins with pure water: CD and NMR studies of protein fragments solubilized in salt-free water. Biophys J. 2006 Dec 1;91(11):4201-9. Epub 2006 Sep 15 PubMed.
- Lim L, Lee X, Song J. Mechanism for transforming cytosolic SOD1 into integral membrane proteins of organelles by ALS-causing mutations. Biochim Biophys Acta. 2015 Jan;1848(1 Pt A):1-7. Epub 2014 Oct 12 PubMed.
- Qin H, Lim L, Wei Y, Gupta G, Song J. Resolving the paradox for protein aggregation diseases: a common mechanism for aggregated proteins to initially attack membranes without needing aggregates. F1000Res. 2013;2:221. Epub 2013 Oct 21 PubMed.
- Polymenidou M, Cleveland DW. The seeds of neurodegeneration: prion-like spreading in ALS. Cell. 2011 Oct 28;147(3):498-508. PubMed.
- Murakami T, Qamar S, Lin JQ, Schierle GS, Rees E, Miyashita A, Costa AR, Dodd RB, Chan FT, Michel CH, Kronenberg-Versteeg D, Li Y, Yang SP, Wakutani Y, Meadows W, Ferry RR, Dong L, Tartaglia GG, Favrin G, Lin WL, Dickson DW, Zhen M, Ron D, Schmitt-Ulms G, Fraser PE, Shneider NA, Holt C, Vendruscolo M, Kaminski CF, St George-Hyslop P. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron. 2015 Nov 18;88(4):678-90. Epub 2015 Oct 29 PubMed.
- Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, McKnight SL. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012 May 11;149(4):768-79. PubMed.
- Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, Grishin NV, Frantz DE, Schneider JW, Chen S, Li L, Sawaya MR, Eisenberg D, Tycko R, McKnight SL. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012 May 11;149(4):753-67. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Qin H, Lim LZ, Wei Y, Song J. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc Natl Acad Sci U S A. 2014 Dec 30;111(52):18619-24. Epub 2014 Dec 12 PubMed.
- Budini M, Romano V, Quadri Z, Buratti E, Baralle FE. TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum Mol Genet. 2015 Jan 1;24(1):9-20. Epub 2014 Aug 13 PubMed.
Primary Papers
- Lim L, Wei Y, Lu Y, Song J. ALS-Causing Mutations Significantly Perturb the Self-Assembly and Interaction with Nucleic Acid of the Intrinsically Disordered Prion-Like Domain of TDP-43. PLoS Biol. 2016 Jan;14(1):e1002338. Epub 2016 Jan 6 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.