Gene Expression Map of Human Body Gives Value to Variants
Quick Links
The DNA packaged neatly inside the nucleus of almost every cell in a person may be identical, but which parts are translated varies dramatically by cell type and by individual genome. In a Herculean effort to connect form to function, the Genotype-Tissue Expression (GTEx) project correlated genetic variations in 449 people with gene expression patterns among 44 postmortem tissue samples, including 10 regions in the brain. Published in four papers in the October 11 Nature, the data present a bird’s-eye view of the diversity of gene regulation across the human body and across the human population. While some genetic variations exerted similar sway over gene expression in most organs, others modulated transcription in specific tissues. The landscape of gene regulation in the brain emerged as strikingly distinctive from other regions of the body.
- GTEx married genomics with transcriptomics in 44 human tissues taken from 449 donors.
- They identified thousands of variants that controlled expression of nearby genes, and genes on other chromosomes.
- Researchers can use GTEx to ask whether a GWAS hit exerts influence over gene expression in the brain and/or elsewhere in the body.
“Frankly, this is a landmark piece of work,” commented John Hardy of University College London. “It provides an amazing resource for all of biology.”
Among other uses, GTEx will allow neurodegenerative disease researchers to investigate whether disease risk variants identified in genome-wide association studies (GWAS) actually trigger meaningful gene expression changes in the brain or elsewhere, commented Philip De Jager of Columbia University in New York. GTEx stands out in its attempt to look at as many tissues as possible within each donor. “That was a visionary decision they made,” De Jager told Alzforum.
Large-scale GWAS and sequencing studies have linked genetic variations to disease risk, but how many of those variants work remains a mystery, especially since most of them land within the abyss of non-coding regions of the genome. If and how such variations influence expression of nearby genes, and whether they do so in the brain, are key questions for researchers. Answers may come from searching for expression quantitative trait loci (eQTL)—genetic variations that influence gene expression. These eQTLs vary among tissues, and several recent studies, including one headed by De Jager, have examined them in the brain, finding that a sizable proportion of GWAS hits also happened to influence gene expression of nearby genes (Sep 2014 news; Fromer et al., 2016; Ng et al., 2017).
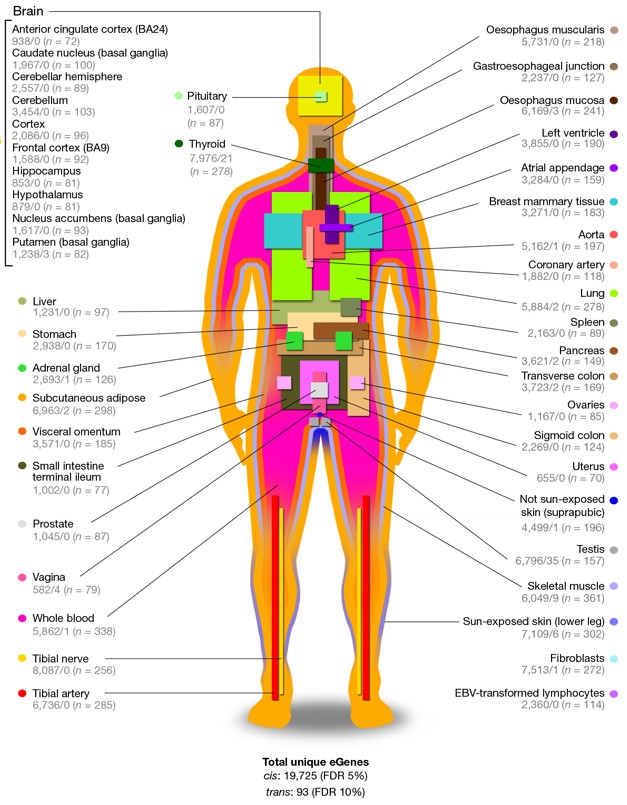
Human-wide Map. Forty-four human tissues sampled for GTEx, listing the number of genes influenced by variants nearby/more than 1Mb away, and the number of samples (n). [Courtesy of GTEx Consortium, Nature 2017.]
The GTEx Consortium moved beyond the cranium, mapping the eQTL landscape across the human body. Infused with resources from the NIH Common Fund in 2010, the project ran an initial pilot phase to streamline collection and processing of postmortem tissue samples, run genetic analyses, and present the data in an interactive portal (GTEx Consortium 2013, 2015). The Broad Institute of MIT and Harvard serves as the project’s central command center, where a team of researchers led by Kristin Ardlie directs the processing and analysis of samples. The specimens come from dozens of sources, including various organ donor programs run by the National Disease Research Interchange and Roswell Park Cancer Institute. Around 40 principle investigators, and many more researchers, are part of the consortium. The current studies include data from more than 7,000 tissue samples gathered postmortem from 449 donors, including just over 100 brain donors. Forty-four distinct tissues, each represented by at least 70 different donors and deemed “healthy” by three independent histologists, were assessed. These tissues included 10 separate regions in the brain. Ultimately, the consortium aims to acquire 17,000 samples from 1,000 donors, according to Stephen Montgomery of Stanford University in Palo Alto, California, a GTEx consortium investigator and co-corresponding author on two of the current Nature papers. RNA sequencing, extensive genotyping, and in some cases whole-genome or exome sequencing, allows GTEx scientists to draw correlations between DNA sequence and gene expression.
One of the GTEx papers presents a high-level view of the eQTL landscape across the samples. Researchers led by co-corresponding authors Montgomery, Alexis Battle of Johns Hopkins University in Baltimore, Christopher Brown of the University of Pennsylvania in Philadelphia, and Barbara Engelhardt of Princeton University in New Jersey identified 152,869 eQTLs that reside within 1Mb of target gene start sites. These cis-eQTLs control nearly 20,000 genes, representing about 80 percent of all protein-coding genes, and half of all long intergenic noncoding (linc) RNAs. They also struck a distinctively bimodal pattern: They acted either broadly across tissues, or specifically in a small subset of highly related tissues. Compared with other related tissues in the body, regions within the brain had the highest proportion of cis-eQTLs in common, and the brain as a whole stood out as most distinctive in its eQTL repertoire. The researchers also searched for trans-eQTLs. These control genes from afar, sometimes even on different chromosomes. Classic examples are variations that alter the expression of transcription factors, which then control myriad other genes. Owing to the distance from their targets, trans-eQTLs require about triple the statistical power to fish out compared to cis-eQTLs, Montgomery told Alzforum. Within 16 tissue types with enough samples for this stringent analysis, the researchers identified 673 trans-eQTLs controlling 112 loci and 93 genes. A majority of trans-eQTLs regulated genes in only one tissue, or a subset of related tissues. Others were more promiscuous. Within the brain, the researchers identified trans-eQTLs only within the putamen, where 11 variants influenced expression of three faraway genes: RMDN3, SEPN1, and CSRNP3. The testis was the runaway winner, sporting 267 trans-eQTLs influencing expression of 35 genes.
How did the eQTLs identified in GTEx relate to disease-associated variants from GWAS? For one thing, tissue-specific eQTLs were more likely to change the expression of disease-related genes than were eQTLs shared broadly among tissues. Considering summary statistics from multiple GWAS, the researchers found that just over half of trait-associated variants matched up with a GTEx eQTL in at least one tissue. However, the relationships are far from simple. For example, about half of all variants that were both a GWAS hit and an eQTL appeared to influence the expression of multiple genes, and did so in an average of four tissues.
Nonetheless, the GTEx eQTL data offers researchers a way to prioritize GWAS hits, De Jager told Alzforum. For example, when he and colleagues gave more weight in a schizophrenia GWAS to hits that were also eQTLs, a new batch of previously insignificant variants rose to their attention.
Mark Cookson of the National Institutes of Health in Bethesda, Maryland, agreed that prioritizing GWAS hits that are also eQTLs is one way to filter through massive data sets, but he cautioned that this approach is imperfect. “Sometimes a variant might affect disease risk, but have no obvious effect on gene expression, and we don’t know why,” he told Alzforum. He added that the next important step will be to connect the dots between variation and gene expression by adding layers of epigenomic data to the eQTL analyses. This could reveal specific mechanisms that drive changes in expression of target genes.
De Jager and others carry out such analyses. Focusing on the prefrontal cortex, his group has identified methylation (m)QTLs and histone acetylation (ha)QTLs, which affect the epigenetic modifications that silence or promote gene expression (Ng et al., 2017). They then asked how many eQTLs were also epigenetic QTLs. For example, a variant that enhances expression of a gene might be expected to also promote histone acetylation in or around that gene. Surprisingly, De Jager told Alzforum that perfect matches between effects on expression and epigenetic marks rarely occurred. “For example, there are many instances where a variant alters epigenetic marks around a target gene, but not the gene’s transcription.” Clearly much is left to learn, he said.
Notably, a plan to add these additional layers of analyses to existing and future GTEx samples is already underway. As described in a separate commentary in Nature, the Enhancing GTEx (EGTEx) project will look at various factors beyond the transcriptome, including epigenetic modifications, chromatin accessibility, post-transcriptional modifications, telomere length, and protein abundance (see image below).

Enhancing GTEx. This consortium will add multiple layers of analysis to GTEx samples, to allow a more in-depth look at how variations affect gene expression. [Courtesy of eGTEx Project, Nature 2017.]
While GWAS primarily served to reveal common variants, whole-genome and exome sequencing studies are beginning to unearth rare variants. These tend to affect disease risk more strongly than their common counterparts. In the second Nature paper to come out of the GTEx Consortium, Battle, Montgomery and colleagues asked whether rare variants exerted their strong effects by altering gene expression. First, they identified genomic outliers—genes expressed at least 10 times more or less robustly than average—among all the GTEx samples. They found that the 449 tissue donors had a median of 83 gene expression outliers per tissue, as well as 10 genes expressed at extreme highs or lows in multiple tissues. Strikingly, these genes tended to have rare variants nearby. Compared to just 8 percent of non-outlier genes, 58 percent of underexpression and 28 percent of overexpression outliers had a rare variant in their vicinity. These tended to be insertions or deletions rather than single nucleotide substitutions, and were more strongly tied to multi-tissue outliers than tissue-specific ones. Whether the rare variants cause the unusual robust or weak expression of the outlier genes remains to be seen.
Commentators agreed that the GTEx data, which has been openly available from the beginning, will be highly useful to neurodegenerative disease researchers. For one thing, Cookson said the eQTL findings lend “directionality” to GWAS hits. “If we can infer from an eQTL analysis that higher expression of a gene is bad, then we’ll know that we need to antagonize the expression of that gene or others in its network,” Cookson said. “From a drug development standpoint, that’s huge.”
Carlos Cruchaga of Washington University in St. Louis had been tapping the GTEx data long before the current publications. He praised the GTEx website for its ease of use, explaining that registered researchers can enter their favorite genes or variants and navigate corresponding eQTLs throughout the body and brain. Though other brain eQTL studies, such as De Jager’s, have used more brain samples than GTEx has so far, Cruchaga pointed out that GTEx’s breadth of non-brain tissues provides a vital resource for the neurodegenerative disease community. For example, researchers can use immune cells outside of the brain as proxies for those within the brain. This is crucial because as of now, brain eQTL maps do not distinguish between different cell types in the brain, such as neurons and microglia. Until single-cell eQTL analyses are conducted, researchers can at least look to related, circulating immune cells such as macrophages as proxies for microglial expression patterns, Cruchaga said.
Montgomery added that given the increasingly recognized importance of systemic inflammation in neurodegenerative disease, considering eQTLs in circulating cells creates a more complete picture of the disease landscape. Case in point, researchers led by Alice Chen-Plotkin at the University of Pennsylvania in Philadelphia used GTEx data to sort out how a common variant associated with the TMEM106b gene boosted risk for frontotemporal lobar degeneration (FTLD). They nailed down a single variant that promoted expression of this gene both in the brain and in immune cells, and went on to uncover the epigenetic mechanism involved. That similar regulatory mechanisms were afoot in the brain and immune cells could allow researchers to use the latter as a window to the regulatory changes that contribute to FTLD and other neurodegenerative diseases.
In addition to the eQTL overview and the analysis of rare variants, two other GTEx papers appeared in the same issue of Nature. One focused on X-chromosome inactivation, the process that silences one of the two copies of X chromosomes in females. They found that nearly a quarter of genes on the chromosome escape inactivation, providing a basis for sex-specific differences in gene expression. In another paper, researchers examined the landscape of RNA editing—a post-translational mechanism that changes adenosine to inositol. They found that editing levels varied between organs, especially in non-repetitive stretches of sequence.—Jessica Shugart
References
News Citations
Paper Citations
- Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, Ruderfer DM, Oh EC, Topol A, Shah HR, Klei LL, Kramer R, Pinto D, Gümüş ZH, Cicek AE, Dang KK, Browne A, Lu C, Xie L, Readhead B, Stahl EA, Xiao J, Parvizi M, Hamamsy T, Fullard JF, Wang YC, Mahajan MC, Derry JM, Dudley JT, Hemby SE, Logsdon BA, Talbot K, Raj T, Bennett DA, De Jager PL, Zhu J, Zhang B, Sullivan PF, Chess A, Purcell SM, Shinobu LA, Mangravite LM, Toyoshiba H, Gur RE, Hahn CG, Lewis DA, Haroutunian V, Peters MA, Lipska BK, Buxbaum JD, Schadt EE, Hirai K, Roeder K, Brennand KJ, Katsanis N, Domenici E, Devlin B, Sklar P. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016 Nov;19(11):1442-1453. Epub 2016 Sep 26 PubMed.
- Ng B, White CC, Klein HU, Sieberts SK, McCabe C, Patrick E, Xu J, Yu L, Gaiteri C, Bennett DA, Mostafavi S, De Jager PL. An xQTL map integrates the genetic architecture of the human brain's transcriptome and epigenome. Nat Neurosci. 2017 Oct;20(10):1418-1426. Epub 2017 Sep 4 PubMed.
- GTEx Consortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013 Jun;45(6):580-5. PubMed.
- GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015 May 8;348(6235):648-60. Epub 2015 May 7 PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI, Biospecimen Collection Source Site—RPCI, Biospecimen Core Resource—VARI, Brain Bank Repository—University of Miami Brain Endowment Bank, Leidos Biomedical—Project Management, ELSI Study, Genome Browser Data Integration &Visualization—EBI, Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz, Lead analysts:, Laboratory, Data Analysis &Coordinating Center (LDACC):, NIH program management:, Biospecimen collection:, Pathology:, eQTL manuscript working group:, Battle A, Brown CD, Engelhardt BE, Montgomery SB. Genetic effects on gene expression across human tissues. Nature. 2017 Oct 11;550(7675):204-213. PubMed.
- Li X, Kim Y, Tsang EK, Davis JR, Damani FN, Chiang C, Hess GT, Zappala Z, Strober BJ, Scott AJ, Li A, Ganna A, Bassik MC, Merker JD, GTEx Consortium, Laboratory, Data Analysis &Coordinating Center (LDACC)—Analysis Working Group, Statistical Methods groups—Analysis Working Group, Enhancing GTEx (eGTEx) groups, NIH Common Fund, NIH/NCI, NIH/NHGRI, NIH/NIMH, NIH/NIDA, Biospecimen Collection Source Site—NDRI, Biospecimen Collection Source Site—RPCI, Biospecimen Core Resource—VARI, Brain Bank Repository—University of Miami Brain Endowment Bank, Leidos Biomedical—Project Management, ELSI Study, Genome Browser Data Integration &Visualization—EBI, Genome Browser Data Integration &Visualization—UCSC Genomics Institute, University of California Santa Cruz, Hall IM, Battle A, Montgomery SB. The impact of rare variation on gene expression across tissues. Nature. 2017 Oct 11;550(7675):239-243. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.