Familial ALS Linked to Both Neuron and Astrocyte Pathology
Quick Links
Animal models and engineered cells have contributed their fair share of insight into the pathogenesis of amyotrophic lateral sclerosis (ALS), but researchers do not often have the opportunity to peer into afflicted human neurons. In a study published May 30 in Cell Reports, researchers led by Rickie Patani and Sonia Gandhi at University College London offer a new model for human disease by using motoneurons and astrocytes derived from amyotrophic lateral sclerosis (ALS) patient stem cells.
The researchers generated spinal motoneurons and astrocytes from induced pluripotent stem cells (iPSCs) of ALS patients carrying mutations in the valosin-containing protein (VCP) gene. VCP mutations account for 2 percent of familial ALS cases and have been linked to other disorders as well, including hereditary inclusion body myopathy with Paget disease of bone and frontotemporal dementia (IBMPFD), jointly termed multisystem proteinopathy.
Previously, researchers reported that VCP mutations promoted the formation of cytoplasmic aggregates of TDP-43, one of the hallmarks of motoneuron disease, and that these aggregates were at least partially responsible for the VCP-induced neurodegeneration seen in a fly model (Neumann et al., 2007; Ritson et al., 2010).
Few studies have tested what VCP mutations do to human neurons, however. Using an iPSC-derived neuronal model, Virginia Kimonis at the University of California, Irvine, found that the VCP R155H mutation boosted cells’ levels of TDP-43 and several proteins involved in protein disposal (Dec et al., 2014).
In search of a more complete view of how VCP’s effects unfold over time, first co-authors Claire Hall, Zhi Yao, Minee Choi, and Giulia Tyzach used iPSC-derived populations of spinal cord motoneurons and astrocytes to track several cellular functions. The cells originated from two patients (four clones) with VCP mutations and three healthy controls.

Nuclear Leak.
TDP-43 (red) stays in the nucleus (blue) in control motoneurons (left) but leaks into the cytoplasm in three-day old VCP-mutant motoneurons (right). [Cell Reports, Hall et al., 2017.]
More VCP-mutant motoneurons than controls died; they also made fewer synapses with their neighbors, and had less intense and fewer coordinated bursts of firing than controls. Monitoring the whereabouts of TDP-43, the authors found no difference between mutant and normal cells when the cells were at an early stage of differentiation, as neural precursor cells. But by the third day after becoming motoneuron-like, the mutant cells were leaking TDP-43 from their nuclei into the cytoplasm (see image above). At the same time, they had increased levels of markers of endoplasmic reticulum (ER) stress, became less able than controls to survive an ER stress assay, and began producing reactive oxygen species (ROS) at a higher rate.
By day 17, the mutant motoneurons had sickly ER tubules. Some were swollen, others cozied up to mitochondria, both signs of continued ER stress. The neurons also had low mitochondrial membrane potential and low glutathione levels, indicating mitochondrial dysfunction and oxidative stress, respectively.
Putting these findings together, Patani and Gandhi suggested that “cyotosolic TDP-43 generates ER stress, which next triggers increased tethering of the ER to mitochondria.” This could depolarize mitochondria, which could impair mitochondrial function and ultimately lead to high levels of free radical production and oxidative stress. “The findings support a role for TDP-43 wreaking havoc in the cytoplasm,” said Patani.
The mechanism that links TDP-43 and ER stress remains uncertain, said Kimonis. Patani and Gandhi acknowledge the link is probably complex, noting that, in addition to cyotplasmic TDP-43 aggregates inducing ER stress, ER stress itself seems to drive TDP-43 leakage (Walker et al., 2013).
The researchers then probed how mutant VCP might affect astrocytes. Studies of astrocytes in other ALS models have yielded varying results depending on the mutation. Superoxide dismutase 1 (SOD1) mutations, for example, can turn astrocytes into motoneuron killers (see Oct 2011 news), but it is unclear if they endanger the survival of the astrocytes themselves. Conversely, mutations in TDP-43 seem to sicken astrocytes without making them toxic to neighboring neurons (Serio et al., 2013).
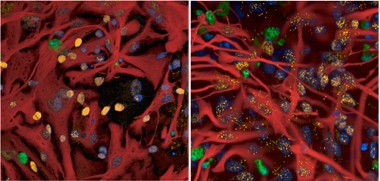
Thanks for nothing, astrocyte.
On the left, healthy astrocytes (red) protect VCP-mutant motoneurons (yellow) from death (green). VCP-mutant astrocytes are much less protective (right). [Courtesy of Zhi Yao.]
Interestingly, Patani and Gandhi found that VCP mutations had both cell-autonomous and non-cell-autonomous effects. They increased astrocytes’ risk of death and rendered them less able to support the survival of both control and VCP-mutant motoneurons (see image at right). The authors also found that the cell-autonomous effects on astrocytes differed from those in motoneurons. VCP-mutant astrocytes seemed more resistant when challenged with an ER stressor and had only a transient drop in mitochondrial membrane potential and a transient increase in ROS, with no change in glutathione levels.
“The study is a validation of key prior findings using iPS-derived neurons and glia from patients, which is a nice advance,” wrote Paul Taylor at St. Jude Children’s Research Hospital in Memphis, Tennessee. “The characterization of the role of glia is also interesting and potentially important.”
Looking ahead, Patani and Gandhi want to generate isogenic controls, that is, cells derived from the same patients but with repaired VCP mutations. They also want to create iPSC-derived upper motoneurons, and to better understand muscle pathology, Kimonis hopes to extend her investigation of iPSC-derived muscle cells (Llewellyn et al., 2017).
Other model systems will be of value, too. iPSC-derived models are not equivalent to the decades-old cells inhabiting an adult nervous system. A study of dopaminergic neurons, for example, revealed that gene expression and DNA methylation patterns differed between iPSC-derived neurons and their in vivo counterparts (Roessler et al., 2014).
In the long term, Patani hopes that shedding light on the sequence of events that marks motoneuron degeneration in human cells will enable researchers to more effectively search for ALS therapies.—Marina Chicurel
References
Alzpedia Citations
News Citations
Paper Citations
- Neumann M, Mackenzie IR, Cairns NJ, Boyer PJ, Markesbery WR, Smith CD, Taylor JP, Kretzschmar HA, Kimonis VE, Forman MS. TDP-43 in the ubiquitin pathology of frontotemporal dementia with VCP gene mutations. J Neuropathol Exp Neurol. 2007 Feb;66(2):152-7. PubMed.
- Ritson GP, Custer SK, Freibaum BD, Guinto JB, Geffel D, Moore J, Tang W, Winton MJ, Neumann M, Trojanowski JQ, Lee VM, Forman MS, Taylor JP. TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J Neurosci. 2010 Jun 2;30(22):7729-39. PubMed.
- Dec E, Ferguson D, Nalbandian A, Gargus M, Katheria V, Rana P, Ibrahim A, Hatch M, Lan M, Llewellyn KJ, Keirstead H, Kimonis VE. Disease-specific Induced Pluripotent Stem Cell Modeling: Insights into the Pathophysiology of Valosin Containing Protein (VCP) Disease. J Stem Cell Res Ther 4:168 2 Feb 2014
- Walker AK, Soo KY, Sundaramoorthy V, Parakh S, Ma Y, Farg MA, Wallace RH, Crouch PJ, Turner BJ, Horne MK, Atkin JD. ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS One. 2013;8(11):e81170. Epub 2013 Nov 29 PubMed.
- Serio A, Bilican B, Barmada SJ, Ando DM, Zhao C, Siller R, Burr K, Haghi G, Story D, Nishimura AL, Carrasco MA, Phatnani HP, Shum C, Wilmut I, Maniatis T, Shaw CE, Finkbeiner S, Chandran S. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc Natl Acad Sci U S A. 2013 Mar 19;110(12):4697-702. PubMed.
- Llewellyn KJ, Nalbandian A, Weiss LN, Chang I, Yu H, Khatib B, Tan B, Scarfone V, Kimonis VE. Myogenic differentiation of VCP disease-induced pluripotent stem cells: A novel platform for drug discovery. PLoS One. 2017;12(6):e0176919. Epub 2017 Jun 2 PubMed.
- Roessler R, Smallwood SA, Veenvliet JV, Pechlivanoglou P, Peng SP, Chakrabarty K, Groot-Koerkamp MJ, Pasterkamp RJ, Wesseling E, Kelsey G, Boddeke E, Smidt MP, Copray S. Detailed analysis of the genetic and epigenetic signatures of iPSC-derived mesodiencephalic dopaminergic neurons. Stem Cell Reports. 2014 Apr 8;2(4):520-33. Epub 2014 Apr 3 PubMed.
Further Reading
Papers
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006 Oct 6;314(5796):130-3. PubMed.
Primary Papers
- Hall CE, Yao Z, Choi M, Tyzack GE, Serio A, Luisier R, Harley J, Preza E, Arber C, Crisp SJ, Watson PM, Kullmann DM, Abramov AY, Wray S, Burley R, Loh SH, Martins LM, Stevens MM, Luscombe NM, Sibley CR, Lakatos A, Ule J, Gandhi S, Patani R. Progressive Motor Neuron Pathology and the Role of Astrocytes in a Human Stem Cell Model of VCP-Related ALS. Cell Rep. 2017 May 30;19(9):1739-1749. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.