In Entorhinal Cortex, Histone “Acetylome” Bears Mark of Alzheimer’s Disease
Quick Links
When it comes to appreciating the depths of genome regulation, reading the genetic code itself is but the tip of the iceberg. Modifications to those DNA sequences, and to the histones they hug, dictate which genes will be expressed when and where. In the October 22 Nature Neuroscience, researchers led by Leonard Schalkwyk at the University of Essex and Jonathan Mill at the University of Exeter, U.K., described a drastically altered histone “acetylome” in postmortem entorhinal cortex samples from people with AD. Looking across the genome, the scientists saw that the extent of H3K27 acetylation—which tends to mark zones of active transcription—varied at more than 4,000 spots scattered throughout, including in areas near key AD genes encoding amyloid precursor protein, presenilin 1 and 2, and tau. It is unclear whether these acetylation changes were a cause or consequence of disease, but the findings demonstrate a strong relationship between the two.
- AD samples contain 4,000 differentially acetylated H3K27 peaks.
- Those peaks stick out near APP, PS1, PS2, MAPT, and other genes tied to AD.
- Only some acetylation differences correlated with altered gene expression.
Some years after genome-wide association studies (GWAS) linked polymorphisms in the genetic code to risk for AD and myriad other diseases, efforts to understand how the far more complex and dynamic epigenetic marks relate to disease risk have begun in earnest. DNA methylation and histone acetylation are the two best-studied types of epigenetic modification. In oversimplified terms, the former correlates with silencing of gene expression, while the latter tends to mark active enhancers and promoters of transcription. Previously, Mill’s group as well as researchers at Rush University in Chicago charted DNA methylation across the genome in brain samples from people with AD and controls, and identified distinct regions of the genome where methylation correlated with AD pathology (Aug 2014 news).
For the current study, first author Sarah Marzi and colleagues investigated whether histone acetylation patterns would also associate with AD. Of the roughly 100 different histone modifications known to affect gene expression, they chose to map acetylation of the lysine 27 residue in histone 3 (H3K27ac), as this adornment is an established marker of active enhancers and promoters (Creyghton et al., 2010). Because the entorhinal cortex (EC) is one of the earliest regions affected by AD pathology, the researchers mapped H3K27 acetylation patterns in EC tissue dissected from 47 postmortem brains. Twenty-four of the donors had died with AD; 23 had little or no signs of AD pathology.
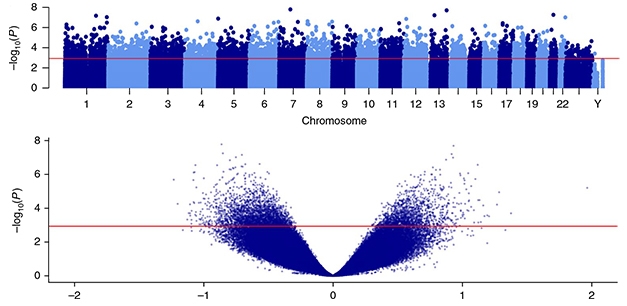
Acetylation Association. Top: A Manhattan plot shows the 4,162 differentially acetylated peaks across all chromosomes in the entorhinal cortices of AD brains, rising above the red line. Bottom: A volcano plot displays the 182,000 H3K27ac peaks detected in entorhinal cortex samples. AD-associated hyper- and hypo-acetylated peaks are above the red line. [Courtesy of Nature Neuro.]
Using chromatin immunoprecipitation-sequencing (CHIP-seq), the researchers identified more than 182,000 H3K27ac peaks across the genome, averaging nearly 1 kilobase in length. Of those peaks, more than 4,000 were differentially acetylated in AD cases. Nearly two-thirds of those AD-associated peaks were hypoacetylated relative to controls; the rest were hyperacetylated. The most strongly hypoacetylated peak rose from the first intron in the ZNF680 gene, which encodes a zinc finger transcription factor expressed in the brain. A peak near both the SOX1 and TEX29 genes—a region known for its brain-specific enhancer activity—was the most hyperacetylated in AD tissue relative to controls.

ZNF680 Hypoacetylated. The most differentially hypoacetylated peak in AD rose from intron 1 of the gene ZNF680. Top: the peak in question is highlighted in vertical pink bar; controls (teal) have higher peaks than AD cases (fuchsia). Below left: acetylation levels in AD versus control brains. [Courtesy of Nature Neuro.]

Among the genes residing near differentially acetylated regions were plenty of AD suspects. MAPT, which encodes tau, had a dramatically hyperacetylated 36kb stretch positioned 10kb upstream of its transcription start site. APP had a hypoacetylated region 100kb downstream of its last exon, while an intron of PSEN1 and a region upstream of PSEN2 featured hyperacetylated peaks.
In addition to these autosomal-dominant AD genes, the researchers also examined whether polymorphisms GWAS have tied to LOAD risk fell within differentially acetylated regions. While they found that more than a third of AD risk polymorphisms overlapped with H3K27 acetylation peaks identified in this study, those hits did not preferentially align with peaks that were differentially acetylated in AD cases.
However, functions of genes near the AD-linked acetylation peaks were enriched for processes relating to AD pathogenesis, including lipoprotein-particle binding, Aβ metabolism, and response to hypoxia.
Did any of these AD-specific acetylation changes echo differences in gene expression? In a larger set of 95 entorhinal cortex samples, including 67 from people with AD, the researchers measured RNA levels of a few select genes that were nearest some of the most differentially acetylated peaks. The results were a mixed bag. For example, expression of RGCC and PIM3, two genes next to hyperacetylated peaks, were elevated in AD cases relative to controls. Expression of GPR22 and KMO, two genes next to hypoacetylated regions, were downregulated in AD cases. So far so good, but not everything lined up in this way. Two other genes near hyperacetylated regions—AKRD17 and ZNF680—were not differentially expressed in AD samples. Muddling matters more, the researchers found that expression of PSEN1 and CR1 were significantly upregulated in AD brain samples, while PSEN2, APP, and MAPT were not differentially expressed.
Mill told Alzforum that he was pleasantly surprised that acetylation differences reflected gene expression in at least some cases. The relationship between histone acetylation and transcription is complex, and active enhancers often turn on distant genes, not their next-door neighbors. “Another possibility is that gene expression can be a much more dynamic, state-dependent outcome than epigenetic changes,” he said. “Once an epigenetic change happens, it might be much more stable, so could reflect something that happened earlier in the disease process,”
Because postmortem studies only inform about a single time point at the end of a decades-long disease process, Mill is using animal models to explore how the relationship between histone acetylation and gene expression changes over time. Such models may allow him to address the question of causality, he said. Mill proposed that some modifications, such as H3K27 acetylation, could instigate disease, while others, such as DNA methylation, might happen in response to pathology. In support of this idea, Mill found almost no overlap between AD-associated changes to DNA methylation and H3K27 acetylation, suggesting the two are governed by different processes.
In a joint comment to Alzforum, Philip Landfield and Eric Blalock of the University of Kentucky College of Medicine in Lexington praised the study, but cautioned that it raised many questions that will need to be addressed before drawing conclusions about the data. “For example, thousands of genes exhibit altered expression with AD, but is histone acetylation closely correlated with these expression changes on a genome-wide basis? In addition, the AD subjects in this study had late-stage AD. Does histone acetylation also distinguish MCI or incipient AD?” they asked. “Answering these difficult questions will require further formidable acetylome-wide analyses, but the present study provides an important initial step toward those answers.”
Even as the relationship between histone acetylation, gene expression, and disease remains poorly understood, histone deacetylase (HDAC) inhibitors, which promote histone acetylation, have shown some promise in animal models of AD (May 2007 news; Dec 2008 news; Cuadrado-Tejedor et al., 2017). Two clinical trials testing HDAC inhibitors in people with AD are underway (clinicaltrials.gov; clinicaltrials.gov). However, two recent studies reported a decrease in HDACs in the brain during AD, although they could not distinguish between different isoforms of the enzyme (Aug 2018 news).
A complementary approach is to directly stoke histone acetylation using histone acetyltransferases. When researchers led by Anne-Laurence Boutillier at the University of Strasbourg, France, tried this to correct flagging histone acetylation in a mouse model of tauopathy, the strategy restored synaptic function and memory (Chatterjee et al., 2018). “Altogether, this gives hope for new therapeutic options targeting the epigenome, especially as Marzi et al. observed a larger number of hypo-acetylated H3K27 peaks than hyper-acetylated ones in human patient’s brains,” Boutillier wrote in a comment to Alzforum.
Mill would not speculate on what his results imply about the therapeutic potential of HDAC inhibition approach. “Our findings provide compelling evidence that histone acetylation is involved in the neurodegenerative process, but it is difficult to say much more at this point.”—Jessica Shugart
References
News Citations
- Alzheimer’s Brains Mottled with Epigenetic Changes
- Memories—Forgotten, But Not Gone?
- DC: Developing But Debatable—Deacetylase Inhibitors for CNS Disease?
- Surprise: HDAC Goes Down, Not Up, in Alzheimer’s Disease
Paper Citations
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010 Dec 14;107(50):21931-6. Epub 2010 Nov 24 PubMed.
- Cuadrado-Tejedor M, Garcia-Barroso C, Sánchez-Arias JA, Rabal O, Pérez-González M, Mederos S, Ugarte A, Franco R, Segura V, Perea G, Oyarzabal J, Garcia-Osta A. A First-in-Class Small-Molecule that Acts as a Dual Inhibitor of HDAC and PDE5 and that Rescues Hippocampal Synaptic Impairment in Alzheimer's Disease Mice. Neuropsychopharmacology. 2017 Jan;42(2):524-539. Epub 2016 Aug 23 PubMed.
- Chatterjee S, Cassel R, Schneider-Anthony A, Merienne K, Cosquer B, Tzeplaeff L, Halder Sinha S, Kumar M, Chaturbedy P, Eswaramoorthy M, Le Gras S, Keime C, Bousiges O, Dutar P, Petsophonsakul P, Rampon C, Cassel JC, Buée L, Blum D, Kundu TK, Boutillier AL. Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Mol Med. 2018 Nov;10(11) PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Marzi SJ, Leung SK, Ribarska T, Hannon E, Smith AR, Pishva E, Poschmann J, Moore K, Troakes C, Al-Sarraj S, Beck S, Newman S, Lunnon K, Schalkwyk LC, Mill J. A histone acetylome-wide association study of Alzheimer's disease identifies disease-associated H3K27ac differences in the entorhinal cortex. Nat Neurosci. 2018 Nov;21(11):1618-1627. Epub 2018 Oct 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UMR7364 CNRS UNISTRA
An important question in current neuroepigenetics research is to understand whether epigenetics is a cause or a consequence of neurodegenerative diseases. Marzi et al. have led a thorough study to tackle the former hypothesis in Alzheimer’s disease by investigating the epigenetic profile of H3K27ac, a mark of active enhancers (Creyghton et al., 2010; Rada-Iglesias et al., 2011; Zentner et al., 2011) in postmortem AD patient’s brains. They led chromatin immunoprecipitation followed by massive parallel sequencing analyses in the entorhinal cortices of 24 AD patients and 23 age-matched low pathology controls. Out of the 4,162 peaks found differentially acetylated between AD and controls, 2,687 were hypo-acetylated and 1,475 were hyper-acetylated.
A strong result from this study is that the authors demonstrated a significant association of differential H3K27ac peaks with regions in proximity of familial AD genes (APP, PSEN1, PSEN2), the MAPT locus, as well as genomic regions containing GWAS variants or genetic risk factors (e.g. APOE) associated with late-onset AD. Together these results could speak to this mark as a potential cause of AD onset. However, there was no consensus on whether the differentially acetylated H3K27 mark was preferentially hyper- or hypo-acetylated when associated with a given AD-related gene, nor a clear association with deregulation of their gene transcription.
A strength of the study is the number of patients investigated. It is noteworthy that histone acetylation modifications are not stable in postmortem tissues and display a great deal variation depending of the postmortem delay (Barrachina et al., 2012), and that the level of total histone is also modified in some brain regions (Narayan et al., 2015). Therefore, it is of most importance to have sufficient statistical power when leading such studies, as was the case in Marzi et al.
Many questions remain unanswered. Analyses were performed in bulk entorhinal tissue, which can lead to confounding effects with regard to which cell type actually displays hyper- or hypo-acetylated H3K27 enrichment. For example, acetylated H3K27 levels have been consistently found enriched in immune and stimulus-response functions enhancers and promoters and decreased in synapse and learning-associated functions enhancers and promoters in an AD mouse model (Gjoneska et al., 2015). Interestingly, in Marzi’s study, the functional analyses led with GREAT generated the interesting result that hypo-acetylated H3K27 peaks were associated with synaptic genes (e.g. neurotransmission, glutamatergic and GABAergic signaling pathways) and this was not the case with hyper-acetylated peaks.
This is reminiscent of our studies investigating the epigenome of affected brain tissues of mice modelling neurodegenerative diseases, including a mouse model of tauopathy (Chatterjee et al., 2018) and one of Huntington’s disease (Achour 2015; Le Gras 2017). In our very recent study, in addition to H3K27ac, we investigated H2Bac, showing it was decreased from early pathological stage in the hippocampus of tauopathic mice. Interestingly, H2Bac peaks colocalized with subsets of H3K27ac peaks, the latter mark being stable between tau and wild-type mice at this age. The hypo-acetylated genomic regions were enriched in genes involved in synaptic functions and regulatory signaling pathways (cAMP, calcium, MAPK …). Our interpretation was that H2Bac decreased levels were a molecular correlate of decreased plasticity in the Tau mice, thus, reflecting epigenetic dysregulation as a consequence of AD pathology.
In HD mice, H3K27 was found significantly hypo-acetylated at neuronal markers. Given that H3K27ac is also enriched at cell-/tissue-specific gene bodies (Hnisz et al., 2013; Whyte et al., 2013), this possibly translates a loss of striatal neuronal identity during HD pathology (Achour et al., 2015). Interestingly, we were able to restore the H2B acetylome in the hippocampi of tau mice treated with a molecule activating CBP/p300 (KAT3A/B) acetyltransferases (Chatterjee et al., 2013), a treatment which also restored LTD, dendritic spine formation, and long-term memory in tau mice (Chatterjee et al., 2018). Altogether, this gives hope for new therapeutic options targeting the epigenome, especially as Marzi et al. observed a larger number of hypo-acetylated H3K27 peaks than hyper-acetylated ones in human patient’s brains.
Important challenges for the future will be to be able to decipher the epigenome in specific cell types (or at least neurons versus non-neuronal cells; e.g. Benito et al., 2015) and to associate chromosome conformation studies, in order to properly understand functional repercussions of deregulated epigenetics at these specific sites found in the vicinity of AD or disease-associated genes as described here by Marzi et al.
References:
Achour M, Le Gras S, Keime C, Parmentier F, Lejeune FX, Boutillier AL, Néri C, Davidson I, Merienne K. Neuronal identity genes regulated by super-enhancers are preferentially down-regulated in the striatum of Huntington's disease mice. Hum Mol Genet. 2015 Jun 15;24(12):3481-96. Epub 2015 Mar 17 PubMed.
Barrachina M, Moreno J, Villar-Menéndez I, Juvés S, Ferrer I. Histone tail acetylation in brain occurs in an unpredictable fashion after death. Cell Tissue Bank. 2012 Dec;13(4):597-606. Epub 2011 Sep 16 PubMed.
Benito E, Urbanke H, Ramachandran B, Barth J, Halder R, Awasthi A, Jain G, Capece V, Burkhardt S, Navarro-Sala M, Nagarajan S, Schütz AL, Johnsen SA, Bonn S, Lührmann R, Dean C, Fischer A. HDAC inhibitor-dependent transcriptome and memory reinstatement in cognitive decline models. J Clin Invest. 2015 Sep;125(9):3572-84. Epub 2015 Aug 17 PubMed.
Chatterjee S, Cassel R, Schneider-Anthony A, Merienne K, Cosquer B, Tzeplaeff L, Halder Sinha S, Kumar M, Chaturbedy P, Eswaramoorthy M, Le Gras S, Keime C, Bousiges O, Dutar P, Petsophonsakul P, Rampon C, Cassel JC, Buée L, Blum D, Kundu TK, Boutillier AL. Reinstating plasticity and memory in a tauopathy mouse model with an acetyltransferase activator. EMBO Mol Med. 2018 Nov;10(11) PubMed.
Chatterjee S, Mizar P, Cassel R, Neidl R, Selvi BR, Mohankrishna DV, Vedamurthy BM, Schneider A, Bousiges O, Mathis C, Cassel JC, Eswaramoorthy M, Kundu TK, Boutillier AL. A novel activator of CBP/p300 acetyltransferases promotes neurogenesis and extends memory duration in adult mice. J Neurosci. 2013 Jun 26;33(26):10698-712. PubMed.
Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010 Dec 14;107(50):21931-6. Epub 2010 Nov 24 PubMed.
Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, Kellis M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015 Feb 19;518(7539):365-9. PubMed.
Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013 Nov 7;155(4):934-47. Epub 2013 Oct 10 PubMed.
Le Gras S, Keime C, Anthony A, Lotz C, De Longprez L, Brouillet E, Cassel JC, Boutillier AL, Merienne K. Altered enhancer transcription underlies Huntington's disease striatal transcriptional signature. Sci Rep. 2017 Feb 22;7:42875. PubMed.
Narayan PJ, Lill C, Faull R, Curtis MA, Dragunow M. Increased acetyl and total histone levels in post-mortem Alzheimer's disease brain. Neurobiol Dis. 2015 Feb;74:281-94. Epub 2014 Dec 5 PubMed.
Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011 Feb 10;470(7333):279-83. Epub 2010 Dec 15 PubMed.
Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013 Apr 11;153(2):307-19. PubMed.
Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res. 2011 Aug;21(8):1273-83. Epub 2011 Jun 1 PubMed.
Make a Comment
To make a comment you must login or register.