Double Whammy: APP Uppsala Deletion Ups Aβ and Its Aggregation Propensity
Quick Links
Researchers have a new entrant to the pantheon of pathogenic APP mutations. The Uppsala mutation, identified in one Swedish family, is the first known multi-codon deletion in the gene to lead to Alzheimer’s disease. It snips six amino acids, residues 19-24, from the middle of the Aβ peptide. In the August 11 Science Translational Medicine, researchers led by Dag Sehlin and Martin Ingelsson at Uppsala University, Sweden, describe how this deletion alters APP processing, boosting production of the pathogenic AβUpp42 peptide. Moreover, this shortened peptide is far more prone to aggregate than is wild-type Aβ42, resulting in rapid plaque deposition and symptom onset around age 40 in the three known carriers. The researchers previously presented the mutation at the 2019 International Conference on Alzheimer’s and Parkinson’s Diseases in Lisbon, Portugal, but didn’t say what it was (Apr 2019 conference news).
- The six-amino-acid Uppsala deletion in APP spurs Aβ42 production.
- AβUpp42 is short, fibrillizes rapidly, and leads to very early onset AD.
- Fibrils are different than wild-type, barely registering on PET scans.
“This is a thorough and extremely convincing story,” Sangram Sisodia at the University of Chicago wrote to Alzforum, lauding the researchers’ use of multiple state-of-the-art mass spectrometry and analytical techniques to characterize amyloid pathology in the mutation carriers. Robert Vassar of Northwestern University, Chicago, was struck by the way this mutation married increased Aβ production with aggressive aggregation. “In a sense, it is like combining the Swedish mutation that makes APP a better substrate for BACE1 together with the Arctic mutation that increases fibrillogenesis,” he wrote (full comment below).

Nip and Tuck. The Uppsala mutation, circled red, snips out residues 690-695 of APP, just above the plasma membrane and very near the α-cleavage site.
Before Uppsala, there were 31 known pathogenic mutations in APP; most are missense mutations, with the only prior deletion being the Osaka mutation, which snips a single glutamic acid from position 693, or residue 22 of Aβ. This lies within the Uppsala deletion region. Many of these known mutations alter APP processing. Typically, in the amyloidogenic pathway, the protein is cleaved sequentially by β-secretase (BACE1) to leave a C-terminal fragment in the cell membrane, and then γ-secretase to produce Aβ40 or Aβ42. In the non-amyloidogenic pathway, initial cleavage by α-secretase prevents the formation of toxic peptides (see image above).
Second author Vilmantas Giedraitis identified the deletion in two siblings and a cousin in Uppsala. When they came to the university’s memory clinic, all three were symptomatic, having problems with executive function, speaking, and basic math, and they scored from 20 to 22 on the MMSE. They were still in their early 40s. The two siblings had neuropsychiatric symptoms, including apathy and anxiety, but their cousin did not. Brain scans showed typical features of Alzheimer’s in all three, with atrophy in the mediotemporal and frontoparietal regions and reduced FDG uptake in temporal and parietal lobes. However, their amyloid PiB PET scans were only weakly positive (see image below). Cerebrospinal fluid findings were also puzzling, with all three carriers having elevated total tau and p-tau181 typical of AD, but Aβ42 levels similar to those of healthy controls. One of the siblings died at age 49 and donated his brain for autopsy; the other two are still alive.
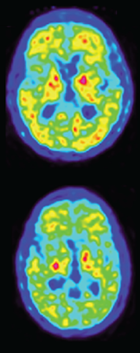
Deceptive PET. Amyloid PiB PET scans of two sibling mutation carriers are only slightly positive, despite a high plaque burden in the brain at autopsy. The top scan came from the sibling with more advanced disease, who passed away. [Courtesy of Pagnon de la Vega et al., Science Translational Medicine/AAAS.]
Despite the CSF and PiB PET data to throw them off track, the researchers suspected some form of early onset AD in the family and sequenced targeted exomes from the three affected members and unaffected relatives. That’s when the APP deletion popped out. It seems to be unique to this family; it has not cropped up in an admittedly small study of around 500 DNA samples from Swedish sporadic AD patients and healthy controls.
To study its effects, first author María Pagnon de la Vega transfected a human kidney cell line with Uppsala APP. Compared to cells transfected with wild-type protein, the Uppsala mutation boosted β-secretase cleavage, and thus Aβ40 and Aβ42 production, by about 50 percent. This high production may explain the CSF Aβ findings, the authors speculated. Aβ normally drops in CSF as the peptide becomes sequestered in plaques in the brain, but high levels of AβUpp42 could mask this effect. Supporting this, only the mutant peptide was high in CSF from the three relatives; wild-type Aβ42 levels were low, similar to those in other AD patients.
In contrast to heightened β-secretase cleavage, no α-secretase cleavage could be detected in these cell cultures, as seen by a lack of the sAPPα fragment. The Uppsala deletion occurs only two amino acids away from the α-secretase cleavage site between amino acids 16 and 17 of Aβ, in effect pulling this site much closer to the cell membrane. This led the authors to wonder if α-cleavage might simply be shifted toward the N-terminus of the protein, further from the membrane. Hinting at this, the authors found high levels of AβUpp5-40 in transfected cultures, indicating the presence of a new cleavage site between residues 4 and 5, 12 amino acids upstream of the normal α-secretase spot. However, it remains to be shown whether α-secretase or some other enzyme creates this fragment. Sisodia suggested that pulse-chase experiments, in which cells briefly take up radiolabeled amino acids, might shed more light on the processing and metabolism of APPUpp.
Not only is more AβUpp made, it also aggregates more readily than wild-type Aβ42. In cell-free assays, synthetic AβUpp42 reached half-maximum fibril concentration in less than an hour, compared to eight hours for wild-type. This is even faster than Aβ42 carrying the Arctic mutation, which takes 1.3 hours. The authors analyzed the two most dominant AβUpp42 fibril species by cryo-electron microscopy (see image below). At 5 Å, the resolution was too low to determine the amino acid sequence, but the shapes of these fibrils roughly resembled previously reported structures (Sep 2015 news; Sep 2017 news). They were not identical, however, suggesting subtle structural differences. Michel Goedert at the MRC Laboratory of Molecular Biology, Cambridge, U.K., noted that the relatively low resolution makes it difficult to compare structures, but the six residues missing from AβUpp42 form the beginning of an S-shaped domain in wild-type fibrils (May 2015 news). Thus, their lack might lead to changes (full comment below).

Fibrils With a New Twist. In vitro, AβUpp42 forms fibrils that roughly resemble wild-type versions, but bend slightly differently. The two most dominant of four polymorphs found are shown. [Courtesy of Pagnon de la Vega et al., Science Translational Medicine/AAAS.]
What happens in the brain? In the brain that came to autopsy, the authors found numerous amyloid plaques throughout all regions, including cerebellum, with the pathology corresponding to Thal stage 5. Mass spectrometry showed that these plaques contained mostly AβUpp42, but also had some AβUpp5-42. If the latter is in fact a product of α-secretase cleavage, that would suggest the non-amyloidogenic pathway is completely abolished by this mutation, with α-cleavage now contributing to aggregation.
With plaque burden high, why is the amyloid PET signal low? The authors speculate that PiB might bind more weakly to AβUpp fibrils than to wild-type, similar to how the Arctic mutation abolishes tracer binding. William Klunk at the University of Pittsburgh suggested another possibility, noting that many early onset AD mutations lead to high amyloid deposition in the basal ganglia, deep in the brain. These deposits tend not to show up on PET scans, which favor the cortical surface. Klunk suggested examining PiB binding to purified AβUpp42 fibrils to resolve this. He also thought the use of cerebellum as a reference region could have clouded the data, given the presence of plaques there, and suggested recalculating the scans using a white-matter reference region (full comment below).
In other respects, the autopsied brain was typical of AD, with tau tangles at Braak stage VI and extensive microgliosis in limbic areas and neocortex. Colin Masters at the University of Melbourne, Australia, noted that like other pathogenic APP mutations, the Uppsala deletion drives tau aggregation, again highlighting the mechanistic connection between the pathologies. Vassar agreed, writing, “This interesting new mutation offers yet additional compelling evidence in favor of the amyloid cascade hypothesis.”
One curiosity, however, was that there were few Aβ oligomers and protofibrils in the Uppsala brain, far less than in most AD brains and instead comparable to the amount found in age-matched healthy controls. Because the mutation carriers developed severe impairment early in life, this may suggest that aggregated forms of AβUpp are highly toxic by themselves, Klunk noted.
The authors plan to study this and other details of APPUpp processing and metabolism in mice. They have generated a model that expresses APP containing the Uppsala and Swedish mutations. They will examine how mutant AβUpp interacts with wild-type, and how this affects aggregation. “We think this might be important for how the amyloid structure forms, and how seeding and propagation occur,” Sehlin told Alzforum.
Meanwhile, Masters believes the mutation highlights the roles of both increased Aβ production and aggregation in disease. “As we enter the era of disease-modifying therapies, the pathogenic mechanisms revealed in this study reinforce the need to develop combination strategies that tackle both Aβ clearance and production,” he wrote to Alzforum (full comment below).—Madolyn Bowman Rogers
References
News Citations
- APP Upp: Mutation Nixes Six Amino Acids from Aβ, Spurs Aggregation
- Electron Microscope Yields Finer Structure of α-Synuclein, Aβ Fibrils
- Amyloid-β Fibril Structure Bares All
- Danger, S-Bends! New Structure for Aβ42 Fibrils Comes into View
Mutation Interactive Images Citations
Mutations Citations
Further Reading
Primary Papers
- Pagnon de la Vega M, Giedraitis V, Michno W, Kilander L, Güner G, Zielinski M, Löwenmark M, Brundin R, Danfors T, Söderberg L, Alafuzoff I, Nilsson LN, Erlandsson A, Willbold D, Müller SA, Schröder GF, Hanrieder J, Lichtenthaler SF, Lannfelt L, Sehlin D, Ingelsson M. The Uppsala APP deletion causes early onset autosomal dominant Alzheimer's disease by altering APP processing and increasing amyloid β fibril formation. Sci Transl Med. 2021 Aug 11;13(606) PubMed. Correction.
Annotate
To make an annotation you must Login or Register.
Comments
University of Pittsburgh
The report by Pagnon de la Vega et al. of a novel APP intra-Aβ deletion brings the opportunity for a deeper understanding of the role of Aβ in pathogenesis of AD. The pattern of PET amyloid tracer retention, in general, and PiB, in particular, has been recognized to be qualitatively and quantitatively different from that in LOAD since our first publication in this area (Klunk et al., 2007).
Depending on the type of mutation, it can be quite predominant in the striatum (as figure 1E suggests). Cortical surface projections such as those shown in figure 1F do not capture this deposition in the basal ganglia and can thus appear only "slightly positive" as described by the authors. In many ADAD mutation types (and Down's syndrome), this may be explained by a predominance of the "cotton-wool" type of amyloid plaque.
However, the Thio-S positivity of the brain examined here suggests otherwise in this case. (By the way, Thio-T and not Thio-S is structurally similar to PiB—although all three, like Congo red, have an affinity for β-sheet fibrils.) One likely issue that may have caused artifactually low apparent PiB retention was the use of the cerebellum as the reference region in this study—as is commonly done in LOAD. It must be assumed that this was the case until proven otherwise by the use of another reference region such as a white-matter area, given that it was stated that, "The regional distribution of Aβ aggregates was extended from neocortex to cerebellum ...", plus the fact that other ADAD mutations have unusually high cerebellar fibrillar amyloid deposition.
Pagnon de la Vega et al. discuss this possibility, but dismiss it due to a low Thio-S-measured burden of cerebellar amyloid in sibling-2. However, the best resolution of this possible artifact would be a recalculation using a white-matter reference. It would also be of great interest to examine the binding of [H-3]PiB to purified fibrils of AβUpp1–42D19–24 and compare the Bmax and affinity to that of Aβwt1-42.
Thus, while the apparently low PiB retention may have a relatively simple methodological explanation, many other interesting findings in this case are more difficult to reconcile with known patterns in LOAD and ADAD. One is the normal (i.e., not reduced) Aβ42 in the CSF. The authors suggest that this is due to overproduction, but could this be due to a normally maintained clearance mechanism in the mutation as well? Other forms of ADAD and Down's syndrome have increased production, but still show decreased CSF Aβ. It did seem like there was relatively little cerebrovascular amyloid in this case and that may also relate to maintenance of normal clearance.
Another is the relative lack of oligomeric forms of AβUpp1–42D19–24 along with the fairly typical—even aggressive—clinical course of the dementia. This suggests the pathogenicity of the fibrillar forms of this peptide is high, although the authors seem to dismiss this without much explanation, as well.
References:
Klunk WE, Price JC, Mathis CA, Tsopelas ND, Lopresti BJ, Ziolko SK, Bi W, Hoge JA, Cohen AD, Ikonomovic MD, Saxton JA, Snitz BE, Pollen DA, Moonis M, Lippa CF, Swearer JM, Johnson KA, Rentz DM, Fischman AJ, Aizenstein HJ, Dekosky ST. Amyloid deposition begins in the striatum of presenilin-1 mutation carriers from two unrelated pedigrees. J Neurosci. 2007 Jun 6;27(23):6174-84. PubMed.
Northwestern University Feinberg School of Medicine
This new, interesting mutation offers yet additional compelling evidence in favor of the amyloid cascade hypothesis.
Interestingly, the Uppsala APP mutation seems to cause AD by both increasing BACE1 cleavage of APP to generate more Aβ, but it also alters the Aβ structure to favor more aggressive Aβ deposition. In a sense, it is like combining the Swedish mutation that makes APP a better substrate for BACE1 together with the Arctic mutation that increases fibrillogenesis—a double whammy that marries increased Aβ production with aggressive aggregation.
That probably explains the exceptionally early age of AD onset of the Uppsala APP mutation carriers. So although this paper does not really shed new conceptual light on the pathogenic mechanism of familial AD, which we already know involves increased Aβ production, Aβ42/40 ratio, or Aβ aggregation, it does strongly support the well-established concept that amyloid accumulation is a critical early step in AD pathogenesis.
MRC Laboratory of Molecular Biology
This interesting paper on a family from Uppsala shows that an intra-Aβ deletion of six amino acids (residues 19-24) caused dominantly inherited Alzheimer’s disease with abundant plaques and tangles. The deletion altered APP processing and increased the assembly of recombinant Aβ42(Δ19-24) into filaments.
As expected, Aβ42 was the major component of Aβ pathology in mutation carriers. Despite the finding that recombinant Aβ1-40(Δ19-24) did not assemble into filaments, some Aβ40 deposits were present in brain. At low pH, recombinant Aβ1-42(Δ19-24) assembled into polymorphic filaments.
Cryo-EM structures of the dominant polymorphs were obtained at 5.7 Å and 5.1 Å resolution. Each polymorph was made of two identical protofilaments, but the low-resolution structures precluded amino acid assignments. It is therefore difficult to compare them with the 4 Å cryo-EM structures of assembled full-length recombinant Aβ(1-42) (Gremer et al., 2017). Residues 19-24 form the beginning of the S-shaped domain that was also observed in the solid-state NMR structures of in vitro aggregated filaments of Aβ42 (Xiao et al., 2015; Colvin et al., 2016; Wälti et al., 2016).
It remains to be seen how the high-resolution structures of Aβ42 filaments from human brain compare with those assembled from recombinant proteins.
References:
Colvin MT, Silvers R, Ni QZ, Can TV, Sergeyev I, Rosay M, Donovan KJ, Michael B, Wall J, Linse S, Griffin RG. Atomic Resolution Structure of Monomorphic Aβ42 Amyloid Fibrils. J Am Chem Soc. 2016 Aug 3;138(30):9663-74. Epub 2016 Jul 14 PubMed.
Gremer L, Schölzel D, Schenk C, Reinartz E, Labahn J, Ravelli RB, Tusche M, Lopez-Iglesias C, Hoyer W, Heise H, Willbold D, Schröder GF. Fibril structure of amyloid-β(1-42) by cryo-electron microscopy. Science. 2017 Oct 6;358(6359):116-119. Epub 2017 Sep 7 PubMed.
Wälti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Böckmann A, Güntert P, Meier BH, Riek R. Atomic-resolution structure of a disease-relevant Aβ(1-42) amyloid fibril. Proc Natl Acad Sci U S A. 2016 Aug 23;113(34):E4976-84. Epub 2016 Jul 28 PubMed.
Xiao Y, Ma B, McElheny D, Parthasarathy S, Long F, Hoshi M, Nussinov R, Ishii Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat Struct Mol Biol. 2015 Jun;22(6):499-505. Epub 2015 May 4 PubMed.
University of Melbourne
This Uppsala deletion is entirely consistent with the general theory that Aβ accumulation causes AD, and that Aβ drives tau aggregation.
As we enter the era of disease-modifying therapies, the pathogenic mechanisms revealed in this study reinforce the need to develop combination strategies that tackle both Aβ clearance and production, either at the same time or sequentially. Passive immunotherapies followed by low-dose BACE1 inhibitors should work in pedigrees such as in this unfortunate family. The tools are available right now. It’s time we applied them.
Co-Director, Brigham and Women's Hospital's Ann Romney Center for Neurologic Diseases
I find this paper quite intriguing, in terms of potentially three mechanisms for the Uppsala mutation's increased Aβ42 accumulation and subsequent downstream AD pathology and clinical features.
The authors did a very nice job teasing out much of the mechanism of this unusual deletion mutation. I am not sure it has direct implications for conventional cases of APP processing and AD development, but it is certainly an interesting and unfortunate error of nature. It does provide another biochemical pathway by which lifelong altered Aβ production and aggregation can produce progressive tauopathy and clinical Alzheimer’s disease.
University of Cincinnati
We would like to highlight a major contradiction between the data and the conclusions in this paper. A key finding is noted as follows: “The amounts of Aβwt1–40 in CSF, produced from their nonmutated APP allele, were lower in patients with the Uppsala APP mutation than in sAD cases and control subjects, whereas Aβwt1–42 was not different in patients with the mutations compared to controls (Fig. 4C)”. However, the data presented in figure 4 clearly shows that the CSF levels of the wild-type (wt), 42 amino acid Aβ42 are significantly lower in mutation carriers compared to controls. As a result, the statement in the Abstract, that “Symptoms and biomarkers are typical of AD, with the exception of normal cerebrospinal fluid (CSF) Aβ42 and only slightly pathological amyloid–positron emission tomography signals,” is incorrect and must be amended. The proper interpretation of the data is critical since this is a new and important mutation that could help understand the pathogenic mechanisms of Alzheimer’s disease.
This clarification is also essential for the discussion on whether Aβ42-related mutations cause Alzheimer's disease via an increase in the soluble Aβ42 to toxic levels, or due to depletion of Aβ42, leading to haploinsufficiency-type loss-of-function. We have recently demonstrated that in amyloid PET-positive individuals, higher levels of soluble Aβ42 in the CSF are associated with normal cognition whereas lower levels are associated with dementia, irrespective of the amyloid plaque load (Sturchio et al., 2021). This is also the case in this report, where mutation carriers with low levels of CSF soluble Aβ42 and nearly normal PET are symptomatic. The data, therefore, support a toxic mechanism for cognitive impairment to be due to Aβ42 depletion rather than its aggregation into plaques (Espay et al, 2021).
References:
Sturchio A, Dwivedi AK, Young CB, Malm T, Marsili L, Sharma JS, Mahajan A, Hill EJ, Andaloussi SE, Poston KL, Manfredsson FP, Schneider LS, Ezzat K, Espay AJ. High cerebrospinal amyloid-β 42 is associated with normal cognition in individuals with brain amyloidosis. EClinicalMedicine, 0, 100988. 2021
Espay AJ, Sturchio A, Schneider LS, Ezzat K. Soluble Amyloid-β Consumption in Alzheimer's Disease. J Alzheimers Dis. 2021;82(4):1403-1415. PubMed.
University of Toronto
Uppsala Universitet
We appreciate the insightful comment by Drs. Ezzat and Espay and are grateful that our colleagues noticed an incorrect statement with respect to the MS-based levels of Aβwt1-42, which were indeed lower in mutation carriers as compared to control subjects. We will seek to have this error corrected in the online version of the paper and/or pointed out as an erratum in the next issue of the journal.
However, we still believe that it is correct to state that mutation carriers had normal CSF levels of Aβ1-42 as we here refer to the ELISA-based AD biomarker. Based on that method, the levels were indeed in the normal range (table S1). However, as the diagnostic ELISA presumably cannot discriminate between the wild-type and the mutated Aβ species (and thus should measure total Aβ1-42), we performed IP-MS which demonstrated that whereas levels of Aβwt1-42 were indeed lower in mutation carriers than in controls, and were similar to those in sporadic AD cases, (fig. 4c), the relative levels of AβUpp1-42Δ19-24 were substantially higher (fig. 4d). When combining these two measurements (to generate total Aβ1-42), the levels were again increased, as opposed to the expected decrease seen for sporadic AD CSF (fig. 4b). Thus, we believe that the more detailed IP-MS analyses offer an explanation as to why the ELISA-based levels of the biomarker Aβ1-42 were not pathologically lowered but were instead in the normal range.
In addition, analyses of post mortem brain tissue from the Uppsala APP mutation carrier revealed high levels of Aβ1-42 both in the TBS and formic acid soluble fractions (figs. 3a-b), suggesting a combination of increased production and deposition in the brain of Aβ1-42. Based on these findings, and on the fact that the mutants were found to display an increased fibrillization rate, we would argue that our data mainly speak in favor of a toxic gain of function for at least this particular APP mutation.
Make a Comment
To make a comment you must login or register.