Does More Aβ38 Mean Less Cognitive Decline in Alzheimer’s?
Quick Links
While clinicians commonly gauge a person's Alzheimer’s disease risk by the amount of Aβ42 in their cerebrospinal fluid, it is unclear exactly what shorter peptide fragments mean in this regard. According to researchers led by Oskar Hansson at Lund University, Sweden, having more Aβ38 protects against AD and slows cognitive decline. In the December 22 Neurology, they report that among people who had early Alzheimer’s as per their CSF phospho-tau181/Aβ42 ratio, those with higher CSF Aβ38 declined less on the Mini-Mental State Exam and were less likely to be diagnosed with dementia four years later. This held true irrespective of APOE genotype, disease stage, or absolute CSF Aβ42 and p-tau181 concentrations. The finding supports the amyloid hypothesis and may buoy efforts to develop modulators of γ-secretase that limit production of Aβ42 in favor of shorter peptides.
- People with high CSF Aβ38 decline more slowly on MMSE.
- This held after correcting for demographics and APOE genotype.
- The finding may rekindle interest in γ-secretase modulators.
“This is a very important story that relates high Aβ38 levels to reduced cognitive decline in two independent cohorts,” wrote Christian Haass of the German Center for Neurodegenerative Diseases, Munich. Although, he added, the mechanism remains unclear. Lucia Chavez Guttierez, KU Leuven, Belgium, offered an idea. “This study supports, from a clinical perspective, the hypothesis that a lower ratio of shorter to longer Aβ peptides plays a role in amyloid toxicity and pathogenicity,” she wrote (full comments below).
Low Aβ42 in the CSF correlates with amyloid plaque load in the brain (Nov 2021 conference news; Oct 2021 news). Being stickier, Aβ42 clumps more easily to form plaques than do shorter Aβ peptides, such as Aβ40 and Aβ38, which tend to remain soluble in the interstitial fluid of the brain. In mice, Aβ40 and Aβ38 only incorporate into plaques that have been initiated by Aβ42 (Jun 2021 news). Given this interplay between Aβ peptides, the researchers wondered how Aβ38 concentration relates to cognitive decline and AD risk.
To find out, first author Nicholas Cullen compared CSF biomarker, demographic, and MMSE data on people whose CSF p-tau181/Aβ42 ratio was above the cutoff for preclinical AD. Of 338 participants in the Swedish BioFINDER study and 318 in ADNI who fit the bill, 71 had reported concerns about memory loss, 342 had mild cognitive impairment, and 243 had been diagnosed with AD dementia.
After controlling for age, sex, years of education, APOE4 genotype, clinical diagnosis, and CSF Aβ42 or p-tau181, the scientists found that higher baseline Aβ38 correlated with slower decline in MMSE score over an average of four years of follow up (see image below). “By including these CSF biomarkers, we adjust the Aβ38 results for the amount of AD pathology in each individual,” Hansson noted.
What explains this correlation between Aβ38 and decline? “Could it be that the higher Aβ38 and Aβ42 values are from subjects with less advanced disease?” wondered Colin Masters, University of Melbourne, Australia (full comment below). In other words, does CSF Aβ38, like Aβ42, simply reflect amyloid plaque load in the brain? Hansson clarified that the correlation between Aβ38 and cognitive decline was independent of disease and people’s CSF Aβ42 level. “The higher a person’s CSF Aβ38, the slower their clinical deterioration regardless of their Aβ42 level,” he told Alzforum.
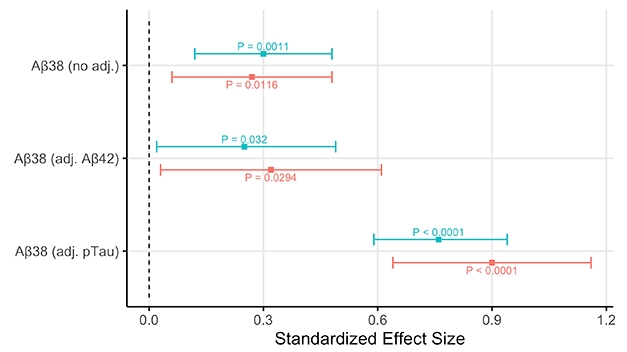
Slow The Slide. In the BioFINDER (blue) and ADNI (red) cohorts, CSF Aβ38 concentration correlated with slower cognitive decline on the MMSE after adjusting for demographics alone (top), demographics and CSF Aβ42 (middle), or demographics and CSF p-tau181 (bottom). [Courtesy of Cullen et al., Neurology, 2021.]
How would CSF Aβ38 relate to the risk of developing full AD dementia? Adjusting for the correlation between CSF p-tau181 and cognitive decline, people in both cohorts whose Aβ38 was high were 45 percent less likely to be clinically diagnosed with dementia on follow up four years later (see image below). This risk remained essentially the same after adjusting for the effects of demographics alone or demographics plus Aβ42 on dementia. The adjusted correlations fit with amyloid/Aβ42 poorly correlating with cognitive decline or dementia, whereas p-tau181 strongly predicts both.

Aβ38 and Dementia. In BioFINDER (blue) and ADNI (red), high CSF Aβ38 correlated with lower risk for dementia after adjusting for demographics and CSF p-tau181 (bottom lines). The correlation was weak after adjusting for only demographics (top), or demographics and Aβ42 (middle). [Courtesy of Cullen et al., Neurology, 2021.]
The researchers also checked how CSF Aβ40 factored in. As with Aβ38, people in both cohorts declined more slowly and were less likely to develop dementia if their baseline Aβ40 levels were high.
All told, the results suggest that people with more of the short Aβ forms in their CSF resist cognitive decline somewhat, despite having AD pathology. Could this have therapeutic implications? The authors suggest γ-secretase modulators to shift Aβ production to less amyloidogenic, smaller fragments, and Bart de Strooper, Dementia Research Institute, London, agreed. “While it is obviously not excluded that Aβ38 has direct protective effects, the more likely explanation is that increased Aβ38 levels reflect a more effective γ-secretase, resulting in less of the long Aβ42 and longer peptides,” he wrote (full comment below).
γ-Secretase modulators (GSMs) can shift Aβ production from Aβ42 toward Aβ38 while keeping the total amount of Aβ steady, skirting off-target effects of inhibiting the enzyme outright (Aug 2013 news; Sep 2011 news; Apr 2011 conference news). Though some are reportedly safe in healthy adults, their efficacy remains unclear as none have reached Phase 3 (Jun 2019 news).
For example, Pfizer’s GSM PF-06648671 shifted Aβ production from longer to shorter peptides while reportedly being safe in multiple Phase 1 studies (Ahn et al., 2020). Still, Agata Copani, University of Catania, Italy, noted the uncertainty of tinkering with the amount of shorter Aβ fragments. “Increasing the concentration of aggregation-prone peptides, as Aβ37 and Aβ38 are, could favor their self-assembly, with unpredictable results,” she wrote (full comment below). Alas, Pfizer halted clinical development of its GSM when it abandoned neurology research in 2018.
Researchers believe these modulators are worth more attention. “It’s my great hope that more than 25 years of fantastic γ-secretase research may not be forgotten, but rather a translational revival may be celebrated,” Haass wrote.—Chelsea Weidman Burke
References
News Citations
- Plasma Aβ—First Sign of AD, But Tough to Measure Prospectively?
- Target or Decoy: Are Drug Developers Chasing the Right Thing?
- iSILK Tracks Growth of Mouse Plaques at Peptide Level
- More Evidence that γ-Secretase Modulators Spare Essential Substrates
- Evidence Mounts That Some γ-Secretase Modulators Bind Presenilin
- Barcelona: Live and Learn—γ-Secretase Inhibitors Fade, Modulators Rise
- γ-Secretase Revisited—Could Selective Inhibitors Treat AD, Cancer Safely?
Therapeutics Citations
Paper Citations
- Ahn JE, Carrieri C, Dela Cruz F, Fullerton T, Hajos-Korcsok E, He P, Kantaridis C, Leurent C, Liu R, Mancuso J, Mendes da Costa L, Qiu R. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin Pharmacol Ther. 2020 Jan;107(1):211-220. Epub 2019 Sep 11 PubMed.
Further Reading
Primary Papers
- Cullen N, Janelidze S, Palmqvist S, Stomrud E, Mattsson-Carlgren N, Hansson O, Alzheimer’s Disease Neuroimaging Initiative. Association of CSF Aβ38 Levels With Risk of Alzheimer Disease-Related Decline. Neurology. 2022 Mar 1;98(9):e958-e967. Epub 2021 Dec 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Gothenburg
These are interesting data from a well-conducted study, particularly in light of it using complementary methods (ELISA and MS). It is therefore of great interest to understand what the changes in CSF Aβ1-38 mean pathologically.
The relevance of higher 1-38 secretion is enigmatic. Cognitively intact subjects (Wiltfang et al., 2002) show generally higher soluble levels of shorter Aβ in CSF, motivated mechanistically by results showing that Aβ1-38 can modulate Aβ1-42 aggregation and mitigate Aβ1-42 neurotoxicity as shown in model systems (Quartey et al., 2021).
Aβ38-mediated Aβ42 pathology should, however, be reflected in a correlation between Aβ38 and Aβ42 as well as Aβ42 correlation with MMSE, respectively, something not observed in the present study of biomarker-positive patients.
The results of this study—that higher Aβ1-38 correlates with higher MMSE after correcting for AD pathology (Aβ1-42 and pTau) and that 1-42 does not—are very interesting. They could indicate, for example, that secretion and deposition of Aβ1-38 itself is of pathogenic relevance. (Similar trends are observed for Aβ1-40.)
In this sense, Aβ1-42 being a general measure for plaque load, Aβ1-38 (and tentatively 1-40) could indicate pathogenic plaque deposition. This aligns pathological studies based on both IHC and our data using novel mass spec imaging approaches, which show that Aβ1-40 and 1-38 are specifically associated with both vascular plaques in CAA as well as cored plaques (Reinert et al., 2014; Michno et al., 2019), while Aβ1-42 was prominent in both diffuse plaques and the diffuse periphery of cored plaques.
Clinically, this can be supported in that Aβ1-38 and Aβ1-40 were associated with steeper cognitive decline than 1-42 (Yu et al., 2019). Further, Aβ1-42 (with its metabolite Aβ4-42) is observed as the sole component in diffuse plaques in patholgical aging, i.e., cognitively normal subjects with plaque pathology who only show diffuse plaques without Aβ1-40 or Aβ1-38 content (Michno et al., 2019).
So, in the presence of AD plaque pathology, higher levels of soluble Aβ1-38 as reflected in CSF, despite significant Aβ plaque burden (estimated by Aβ1-42), could indicate less plaque toxicity, i.e., less vascular and cored/mature plaque pathology. Interestingly, 1-38 secretion and deposition were found to be a secondary event to initial Aβ1-42 production and deposition (Michno et al., 2021) proving that Aβ 1-38 is not a sole metabolite of Aβ1-42. This further highlights that the selective processing of APP into Aβ1-38, and the pathological consequences of that processing, warrants more neuropathological and preclinical/mechanistic studies.
References:
Wiltfang J, Esselmann H, Bibl M, Smirnov A, Otto M, Paul S, Schmidt B, Klafki HW, Maler M, Dyrks T, Bienert M, Beyermann M, Rüther E, Kornhuber J. Highly conserved and disease-specific patterns of carboxyterminally truncated Abeta peptides 1-37/38/39 in addition to 1-40/42 in Alzheimer's disease and in patients with chronic neuroinflammation. J Neurochem. 2002 May;81(3):481-96. PubMed.
Quartey MO, Nyarko JN, Maley JM, Barnes JR, Bolanos MA, Heistad RM, Knudsen KJ, Pennington PR, Buttigieg J, De Carvalho CE, Leary SC, Parsons MP, Mousseau DD. The Aβ(1-38) peptide is a negative regulator of the Aβ(1-42) peptide implicated in Alzheimer disease progression. Sci Rep. 2021 Jan 11;11(1):431. PubMed.
Reinert J, Martens H, Huettenrauch M, Kolbow T, Lannfelt L, Ingelsson M, Paetau A, Verkkoniemi-Ahola A, Bayer TA, Wirths O. Aβ38 in the brains of patients with sporadic and familial Alzheimer's disease and transgenic mouse models. J Alzheimers Dis. 2014;39(4):871-81. PubMed.
Michno W, Nyström S, Wehrli P, Lashley T, Brinkmalm G, Guerard L, Syvänen S, Sehlin D, Kaya I, Brinet D, Nilsson KP, Hammarström P, Blennow K, Zetterberg H, Hanrieder J. Pyroglutamation of amyloid-βx-42 (Aβx-42) followed by Aβ1-40 deposition underlies plaque polymorphism in progressing Alzheimer's disease pathology. J Biol Chem. 2019 Apr 26;294(17):6719-6732. Epub 2019 Feb 27 PubMed.
Yu L, Petyuk VA, Tasaki S, Boyle PA, Gaiteri C, Schneider JA, De Jager PL, Bennett DA. Association of Cortical β-Amyloid Protein in the Absence of Insoluble Deposits With Alzheimer Disease. JAMA Neurol. 2019 Jul 1;76(7):818-826. PubMed.
Michno W, Stringer KM, Enzlein T, Passarelli MK, Escrig S, Vitanova K, Wood J, Blennow K, Zetterberg H, Meibom A, Hopf C, Edwards FA, Hanrieder J. Following spatial Aβ aggregation dynamics in evolving Alzheimer's disease pathology by imaging stable isotope labeling kinetics. Sci Adv. 2021 Jun;7(25) Print 2021 Jun PubMed.
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
I have been waiting for such an analysis for a long time. This is indeed a very important story, which finally relates high Aβ38 levels to reduced cognitive decline in two independent cohorts, although the mechanism remains unclear. Is it a reduction of longer species such as Aβ48, which are very difficult to detect, or does the ratio of short to long peptides affect disease progression via an unknown mechanism?
This finding clearly supports the application of γ-secretase modulators, at least in patients suffering from autosomal-dominant familial Alzheimer’s disease. It also supports the amyloid cascade. It’s my great hope now that more than 25 years of fantastic γ-secretase research may not be forgotten, but rather a translational revival may be celebrated. It is about time!
K.U.Leuven and V.I.B.
This sound study investigated the clinical association between shorter amyloid-β peptides (Aβ38 and Aβ40) and Alzheimer’s disease-related changes in two large, independent cohorts of biomarker-positive individuals who are spread across the AD spectrum. The analyses show that higher CSF Aβ38 levels are associated with less cognitive decline and lower risk of developing AD. This study thus supports, from a clinical perspective, the hypothesis that a lower ratio of shorter to longer Aβ peptides plays a role in amyloid toxicity and pathogenicity.
Studies investigating the relationship between different Aβ peptides and AD have been mainly limited to the analysis of Aβ42 and Aβ40 levels. However, Aβ peptides varying from 37 to 43 amino acids in length are generated physiologically. Notably, shorter peptides, Aβ37 and Aβ38, have been shown to ameliorate Aβ42- and Aβ43-driven toxicity, suggesting a potential protective role for shorter Aβ peptides (Moore et al., 2018). Furthermore, mechanistic analyses of the effects of FAD-linked mutations on APP processing support strategies that shift Aβ production toward shorter Aβ peptides (γ-secretase modulators) as a potential therapeutic avenue to tackle Aβ toxicity (Szaruga et al., 2017).
The clinical study presented here is valuable because it investigates Aβ peptide levels more comprehensively. That the findings demonstrate a protective role for the short Aβ38 peptides in sporadic AD patients is highly relevant. It supports the notion that familial and sporadic AD individuals may both benefit from strategies that modulate Aβ generation to favor the generation of shorter rather than longer Aβ peptides.
References:
Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, Ladd TB, Ran Y, Levites Y, Kukar TL, Kurian JJ, McKenna R, Koo EH, Borchelt DR, Janus C, Rincon-Limas D, Fernandez-Funez P, Golde TE. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018 Jan 2;215(1):283-301. Epub 2017 Dec 5 PubMed.
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-causing mutations shift Aβ length by destabilizing γ-secretase-Aβn interactions. Cell. 2021 Apr 15;184(8):2257-2258. PubMed.
University of Melbourne
The development of biofluid and/or imaging markers for algorithms predicting cognitive decline and disease progression is high on all of our radar screens at the moment. This paper is based on cross-sectional, not longitudinal, CSF biomarkers, with relatively short follow-up times (mean four years) in two cohorts, Biofinder and ADNI, using two separate platforms for CSF Aβ measurements (which is not ideal). Subjects were included if their CSF p-tau/Aβ42 ratios were abnormal, meaning that they met criteria for preclinical (SCI), prodromal (MCI), or clinical (dementia) AD. Use of the p-tau-181/Aβ42 ratio hides from view those outliers with “normal” Aβ42 or p-tau181 levels, yet may represent a subgroup whose trajectories differ from those in the group means. There is currently a lack of consensus in the field as to how to handle these outliers. Our policy is to report all absolute values and the ratio, but also to refer to the ratio with borderline cases (Delaby et al., 2021).
The findings in this cross-sectional study are most interesting. Aβ38 and Aβ42 are in positive equilibrium, both decrease as AD progresses, and p-tau181 also shows a positive relationship with Aβ38 (and presumably with Aβ42, but this data was not shown). The authors report that higher levels of Aβ38 and Aβ42 are associated with slower cognitive decline/disease progression. But the effect sizes are modest (0.3). After adjustment for p-tau181, the effect size becomes moderate (0.8). Could it be that the higher Aβ38 and Aβ42 values are in subjects with less advanced disease? The authors were unable to define this relationship.
The authors also raise the subject of γ-secretase inhibitors (GSI) and modulators (GSM), with the GSMs showing promise to selectively lower Aβ42 while maintaining normal levels of Aβ38 (Rynearson et al., 2021). While GSMs remain to be fully evaluated in human trials, it appears that β-secretase (BACE1) inhibitors can and should be rehabilitated, having been used at dosage levels guaranteed to adversely impact the normal processing of APP (McDade et al., 2021). The same applies to GSIs and presumably to GSMs: Finding the right dose that does not compromise normal APP processing (such as 5-10 percent inhibition) may be more important than whether selective reductions of Aβ42 prevail over Aβ40 and Aβ38.
The torrent of biomarker papers from the Hansson laboratory continues at full pace!
References:
Delaby C, Teunissen CE, Blennow K, Alcolea D, Arisi I, Amar EB, Beaume A, Bedel A, Bellomo G, Bigot-Corbel E, Bjerke M, Blanc-Quintin MC, Boada M, Bousiges O, Chapman MD, DeMarco ML, D'Onofrio M, Dumurgier J, Dufour-Rainfray D, Engelborghs S, Esselmann H, Fogli A, Gabelle A, Galloni E, Gondolf C, Grandhomme F, Grau-Rivera O, Hart M, Ikeuchi T, Jeromin A, Kasuga K, Keshavan A, Khalil M, Körtvelyessy P, Kulczynska-Przybik A, Laplanche JL, Lewczuk P, Li QX, Lleó A, Malaplate C, Marquié M, Masters CL, Mroczko B, Nogueira L, Orellana A, Otto M, Oudart JB, Paquet C, Paoletti FP, Parnetti L, Perret-Liaudet A, Peoc'h K, Poesen K, Puig-Pijoan A, Quadrio I, Quillard-Muraine M, Rucheton B, Schraen S, Schott JM, Shaw LM, Suárez-Calvet M, Tsolaki M, Tumani H, Udeh-Momoh CT, Vaudran L, Verbeek MM, Verde F, Vermunt L, Vogelgsang J, Wiltfang J, Zetterberg H, Lehmann S. Clinical reporting following the quantification of cerebrospinal fluid biomarkers in Alzheimer's disease: An international overview. Alzheimers Dement. 2021 Dec 22; PubMed.
Rynearson KD, Ponnusamy M, Prikhodko O, Xie Y, Zhang C, Nguyen P, Hug B, Sawa M, Becker A, Spencer B, Florio J, Mante M, Salehi B, Arias C, Galasko D, Head BP, Johnson G, Lin JH, Duddy SK, Rissman RA, Mobley WC, Thinakaran G, Tanzi RE, Wagner SL. Preclinical validation of a potent γ-secretase modulator for Alzheimer's disease prevention. J Exp Med. 2021 Apr 5;218(4) PubMed.
McDade E, Voytyuk I, Aisen P, Bateman RJ, Carrillo MC, De Strooper B, Haass C, Reiman EM, Sperling R, Tariot PN, Yan R, Masters CL, Vassar R, Lichtenthaler SF. The case for low-level BACE1 inhibition for the prevention of Alzheimer disease. Nat Rev Neurol. 2021 Nov;17(11):703-714. Epub 2021 Sep 21 PubMed.
UK Dementia Research Institute@UCL and VIB@KuLeuven
One of the few established causes of Alzheimer's disease is the change in the relative ratios of long versus short amyloid peptides in patients with presenilin mutations (Szaruga et al., 2017). Shorter amyloid peptides, for instance Aβ38, reflect effective amyloid precursor protein processing by γ-secretase, while Aβ peptides longer than 41 amino acids are generated if mutations in the presenilin subunit destabilize the enzyme-substrate interaction, resulting in faster release of the substrate, i.e., long Aβ peptides.
Some correlation exists between the age of onset associated with the different presenilin mutations and this phenomenon. Interestingly, Cullen et al. show that increased levels of Aβ38 in CSF are correlated with lower AD-related decline. While it is obviously not excluded that Aβ38 has direct protective effects, the more likely explanation is that increased Aβ38 levels reflect a more effective γ-secretase in these patients, resulting in less of the long (≥42) Aβ peptides. If this is further confirmed, then the result indeed provides a strong argument for the use of γ-secretase modulators. We commented a few years ago that γ-secretase as a drug target was dropped too early (De Strooper, 2014).
References:
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
De Strooper B. Lessons from a failed γ-secretase Alzheimer trial. Cell. 2014 Nov 6;159(4):721-6. PubMed.
University of Catania
Cullen and colleagues have measured CSF Aβ38 levels in 656 individuals across two clinical cohorts. CSF Aβ38 has been measured before and found to be interrelated with Aβ40, as both do not differ significantly between AD patients and cognitively unimpaired subjects. Cullen and colleagues take things a step further, showing that higher CSF Aβ38 levels associate with less cognitive decline and lower risk of developing AD in individuals who are AD-biomarker-positive. Of note, CSF Aβ40 levels show a similar trend.
The first conclusion is that Aβ38 and, possibly, Aβ40 are protective in biomarker-positive individuals. The second conclusion is that the finding renews interest in γ-secretase modulators, which lower Aβ42 but increase the relative production of shorter Aβ fragments (Wagner et al., 2014). Regarding the first point, there is evidence that Aβ38 and Aβ40 may both protect against Aβ42 toxicity in vivo (Moore et al, 2018), even though the mechanism is unknown. The available data are not unequivocal: Aβ38 counteracts Aβ42 toxicity, while triggering the toxicity of Aβ40 (Vandersteen et al., 2012; Quartey et al., 2021). It follows that Aβ38 may not be invariably neuroprotective, rather its role in AD progression requires further elucidation. As with other amyloidogenic peptides, the biological properties of Aβ38 depend on the context, which may or may not facilitate its self-assembly.
Coming to the therapeutic potential of GSMs, it is hard to predict how much benefit AD patients can get from drugs that selectively, or mostly, lower Aβ42. We should recall that the most surprising outcome of the clinical trial with the γ-secretase inhibitor Semagacestat was the worsening of cognitive decline, by some mechanism that remains unexplained (Doody et al., 2013). The possibility that the drug had reduced the physiological monomeric form of Aβ42 to a detrimental level was not considered (Giuffrida et al., 2009; Giuffrida et al., 2015; Santangelo et al., 2021). Yet, cognitive worsening also occurs with BACE1 inhibitors (Egan et al., 2018).
The GSM PF-06648671 has reached Phase 1. In three different studies, the drug has shown a good safety profile after multiple ascending doses for 14 days, and it has shown the expected pharmacodynamics, i.e., reduction of Aβ42 and Aβ40 in CSF, with greater effects on Aβ42, and increase of Aβ37 and Aβ38, with greater effects on Aβ37 (Ahn et al., 2020). Clinical trials with PF-06648671 will demonstrate the therapeutic benefit of lowering longer Aβ species and increasing shorter Aβ fragments. For the latter, there is no proof of lack of toxicity. It might be worth pointing out that increasing the concentration of aggregation-prone proteins, such as Aβ37 and Aβ38, could favor their self-assembly, with unpredictable results.
References:
Wagner SL, Zhang C, Cheng S, Nguyen P, Zhang X, Rynearson KD, Wang R, Li Y, Sisodia SS, Mobley WC, Tanzi RE. Soluble γ-secretase modulators selectively inhibit the production of the 42-amino acid amyloid β peptide variant and augment the production of multiple carboxy-truncated amyloid β species. Biochemistry. 2014 Feb 4;53(4):702-13. Epub 2014 Jan 22 PubMed.
Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, Ladd TB, Ran Y, Levites Y, Kukar TL, Kurian JJ, McKenna R, Koo EH, Borchelt DR, Janus C, Rincon-Limas D, Fernandez-Funez P, Golde TE. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018 Jan 2;215(1):283-301. Epub 2017 Dec 5 PubMed.
Vandersteen A, Masman MF, De Baets G, Jonckheere W, Van Der Werf K, Marrink SJ, Rozenski J, Benilova I, De Strooper B, Subramaniam V, Schymkowitz J, Rousseau F, Broersen K. Molecular Plasticity Regulates Oligomerization and Cytotoxicity of the Multipeptide-length Amyloid-β Peptide Pool. J Biol Chem. 2012 Oct 26;287(44):36732-43. PubMed.
Quartey MO, Nyarko JN, Maley JM, Barnes JR, Bolanos MA, Heistad RM, Knudsen KJ, Pennington PR, Buttigieg J, De Carvalho CE, Leary SC, Parsons MP, Mousseau DD. The Aβ(1-38) peptide is a negative regulator of the Aβ(1-42) peptide implicated in Alzheimer disease progression. Sci Rep. 2021 Jan 11;11(1):431. PubMed.
Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, He F, Sun X, Thomas RG, Aisen PS, , Siemers E, Sethuraman G, Mohs R. A phase 3 trial of semagacestat for treatment of Alzheimer's disease. N Engl J Med. 2013 Jul 25;369(4):341-50. PubMed.
Giuffrida ML, Caraci F, Pignataro B, Cataldo S, De Bona P, Bruno V, Molinaro G, Pappalardo G, Messina A, Palmigiano A, Garozzo D, Nicoletti F, Rizzarelli E, Copani A. Beta-amyloid monomers are neuroprotective. J Neurosci. 2009 Aug 26;29(34):10582-7. PubMed.
Giuffrida ML, Tomasello MF, Pandini G, Caraci F, Battaglia G, Busceti C, Di Pietro P, Pappalardo G, Attanasio F, Chiechio S, Bagnoli S, Nacmias B, Sorbi S, Vigneri R, Rizzarelli E, Nicoletti F, Copani A. Monomeric ß-amyloid interacts with type-1 insulin-like growth factor receptors to provide energy supply to neurons. Front Cell Neurosci. 2015;9:297. Epub 2015 Aug 7 PubMed.
Santangelo R, Giuffrida ML, Satriano C, Tomasello MF, Zimbone S, Copani A. β-amyloid monomers drive up neuronal aerobic glycolysis in response to energy stressors. Aging (Albany NY). 2021 Jul 21;13(14):18033-18050. PubMed.
Egan MF, Kost J, Tariot PN, Aisen PS, Cummings JL, Vellas B, Sur C, Mukai Y, Voss T, Furtek C, Mahoney E, Harper Mozley L, Vandenberghe R, Mo Y, Michelson D. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer's Disease. N Engl J Med. 2018 May 3;378(18):1691-1703. PubMed.
Ahn JE, Carrieri C, Dela Cruz F, Fullerton T, Hajos-Korcsok E, He P, Kantaridis C, Leurent C, Liu R, Mancuso J, Mendes da Costa L, Qiu R. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin Pharmacol Ther. 2020 Jan;107(1):211-220. Epub 2019 Sep 11 PubMed.
Brigham and Women's Hospital
Co-Director, Brigham and Women's Hospital's Ann Romney Center for Neurologic Diseases
This informative new study provides further support for a correlation between altered γ-secretase activity and clinical phenotypes in sporadic AD (SAD) patients. As the final product of the serial tripeptide cleavages of APP in the Aβ48-45-42-38 pathway, higher Aβ38 levels in CSF were found to be associated with less decline in the MMSE in both BioFINDER and ADNI cohorts. It is also interesting that higher CSF Aβ38 levels (putatively beneficial) correlated with higher CSF soluble p-tau levels (usually considered detrimental) in these AD biomarker-positive subjects (Fig. 1C).This is an apparently paradoxical correlation for which the explanatory mechanism remains to be defined. But overall, this study suggests that quantifying γ-secretase “processive” activity in patients entering the SAD trajectory might aid in predicting their future cognitive decline.</p>
During AD pathogenesis, various Aβ levels may be altered in CSF (and plasma) via different mechanisms. Impaired γ-secretase processive cleavage activity would lead to lower Aβ37 and 38 and higher Aβ42 and 43. Aβ42 and 43 may deposit progressively in brain parenchyma, thereby lowering the levels of the free peptides in CSF, while Aβ37 and 38 may not deposit significantly. In PSEN1 and 2 mutant familial AD (FAD) cases, much evidence suggests that a loss of γ-secretase processivity, i.e., the successive carboxypeptidase-like cleavages (“trimming”) to release final catalytic products, is at the root of pathogenesis.
We have recently quantified ratios between short and long Aβs, e.g., Aβ37/40, Aβ37/42, and Aβ38/42, to monitor the severity of impaired γ-secretase processivity by various PSEN1 mutations in cultured cells (Liu et al., 2021). Further, we have measured these “short-long” Aβ ratios in SAD patient-derived iPSC neurons and found the Aβ37/42 ratio to reflect certain phenotypic features, including degree of cognitive decline, of these SAD patients (Lagomarsino et al., 2021). Another recent study found that γ-secretase processive activity (reflected by Aβ38/42 and Aβ40/43 as produced in vitro by γ-secretase extracted from SAD brain tissue) correlated with brain pathology as indicated by Braak senile plaque stages (Kakuda et al., 2020).
In the context of Cullen et al.’s findings, one wonders whether a high Aβ38 level in CSF simply reflects a relatively physiological or healthy γ-secretase processive activity, rather than the high Aβ38 peptide actually conferring a clinical benefit. There is still a lack of evidence to demonstrate cognitive modulatory effects of Aβ38 in vivo, although it may decrease the aggregation of longer Aβs like Aβ42 (Kim et al., 2007).
The presumed mechanism of a GSM is to bind to presenilin in such a way as to allosterically modulate its processive cleavage to reduce longer, more aggregation-prone Aβs, with increased shorter Aβs as a consequence. For example, as shown by three Phase 1 studies, the very potent GSM PF-06648671 induced increases in CSF Aβ37 levels that were much greater than those of Aβ38 (Ahn et al., 2020); this may represent a shared pharmacological effect of non-acidic type GSMs.
Overall, this new study adds to evidence of a potentially shared molecular mechanism, loss of γ-secretase processivity, that may bridge the pathogenesis of FAD and at least some SAD cases. As we discussed previously (Liu et al., 2021), APP is an imperfect substrate for γ-secretase, which produces more proteolytic intermediates (Aβ40 and 42) than final products (Aβ37 and 38). Genetic or age-related biological factors might amplify this intrinsic enzymatic imperfection drastically (in FAD) or mildly (in some cases of SAD).
References:
Liu L, Lauro BM, Wolfe MS, Selkoe DJ. Hydrophilic loop 1 of Presenilin-1 and the APP GxxxG transmembrane motif regulate γ-secretase function in generating Alzheimer-causing Aβ peptides. J Biol Chem. 2021;296:100393. Epub 2021 Feb 8 PubMed.
Lagomarsino VN, Pearse RV 2nd, Liu L, Hsieh YC, Fernandez MA, Vinton EA, Paull D, Felsky D, Tasaki S, Gaiteri C, Vardarajan B, Lee H, Muratore CR, Benoit CR, Chou V, Fancher SB, He A, Merchant JP, Duong DM, Martinez H, Zhou M, Bah F, Vicent MA, Stricker JM, Xu J, Dammer EB, Levey AI, Chibnik LB, Menon V, Seyfried NT, De Jager PL, Noggle S, Selkoe DJ, Bennett DA, Young-Pearse TL. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron. 2021 Nov 3;109(21):3402-3420.e9. Epub 2021 Sep 1 PubMed.
Kakuda N, Yamaguchi H, Akazawa K, Hata S, Suzuki T, Hatsuta H, Murayama S, Funamoto S, Ihara Y. γ-Secretase Activity Is Associated with Braak Senile Plaque Stages. Am J Pathol. 2020 Jun;190(6):1323-1331. Epub 2020 Mar 20 PubMed.
Kim J, Onstead L, Randle S, Price R, Smithson L, Zwizinski C, Dickson DW, Golde T, McGowan E. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007 Jan 17;27(3):627-33. PubMed.
Ahn JE, Carrieri C, Dela Cruz F, Fullerton T, Hajos-Korcsok E, He P, Kantaridis C, Leurent C, Liu R, Mancuso J, Mendes da Costa L, Qiu R. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin Pharmacol Ther. 2020 Jan;107(1):211-220. Epub 2019 Sep 11 PubMed.
French National Centre for Scientific Research
This is an important study that argues for clinical research on γ-secretase modulators. The limiting factor for processing APP-CTFs into Aβ and AICD is not the γ-secretase enzyme but the amount of APP-CTFs substrate (Barthet et al., 2011), and probably the subcellular localization of APP and its secretases, dictating the substrate-enzyme collision.
Under these conditions, stimulating the γ-secretase, i.e., promoting the full processivity of APP cleavage by presenilin, is unlikely to lead to more Aβ but would favor the production of the shortest Aβ species and reduce the abundance of potentially toxic APP-CTFs (Barthet et al., 2012; 2018).
Interestingly, the γ-secretase component APH-1 may represent a druggable target of interest for allosteric modulation of γ-secretase, as its 7-TM structure resembles G protein-coupled receptors, the main drug target class.
References:
Barthet G, Shioi J, Shao Z, Ren Y, Georgakopoulos A, Robakis NK. Inhibitors of γ-secretase stabilize the complex and differentially affect processing of amyloid precursor protein and other substrates. FASEB J. 2011 Sep;25(9):2937-46. PubMed.
Barthet G, Georgakopoulos A, Robakis NK. Cellular mechanisms of γ-secretase substrate selection, processing and toxicity. Prog Neurobiol. 2012 Aug;98(2):166-75. PubMed.
Barthet G, Jordà-Siquier T, Rumi-Masante J, Bernadou F, Müller U, Mulle C. Presenilin-mediated cleavage of APP regulates synaptotagmin-7 and presynaptic plasticity. Nat Commun. 2018 Nov 14;9(1):4780. PubMed.
View all comments by Gael BarthetArkuda Therapeutics
Mutations in presenilin causing familial Alzheimer’s disease reduce γ-secretase activity by way of slower processivity. This, in turn, leads to an increase in the relative ratios of longer (Aβ42, 43) to shorter (Aβ37, 38, 40) Aβ peptides (Szaruga et al., 2017). We and others have shown that γ-secretase modulators (GSMs) can reverse the effect of PS mutations by restoring the activity of GS and shifting Aβ production toward the shorter forms (Blain et al., 2016; Okochi et al., 2013).
This new study by Cullen and colleagues shows that higher levels of CSF Aβ38 correlate with slower cognitive decline across two clinical cohorts. One potential explanation for this effect is that the short forms like Aβ37 and Aβ38 can prevent the aggregation (Blain et al., 2016) and toxicity (Moore et al., 2018) of longer Aβ forms.
This finding should be celebrated and will hopefully revive interest in pursuing GSMs as a therapeutic approach for FAD, now that potent compounds are available for such studies (Bursavich et al., 2021; Ahn et al., 2020).
References:
Szaruga M, Munteanu B, Lismont S, Veugelen S, Horré K, Mercken M, Saido TC, Ryan NS, De Vos T, Savvides SN, Gallardo R, Schymkowitz J, Rousseau F, Fox NC, Hopf C, De Strooper B, Chávez-Gutiérrez L. Alzheimer's-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell. 2017 Jul 27;170(3):443-456.e14. PubMed. Correction.
Blain JF, Bursavich MG, Freeman EA, Hrdlicka LA, Hodgdon HE, Chen T, Costa DE, Harrison BA, Kapadnis S, Murphy DA, Nolan S, Tu Z, Tang C, Burnett DA, Patzke H, Koenig G. Characterization of FRM-36143 as a new γ-secretase modulator for the potential treatment of familial Alzheimer's disease. Alzheimers Res Ther. 2016 Aug 30;8:34. PubMed.
Okochi M, Tagami S, Yanagida K, Takami M, Kodama TS, Mori K, Nakayama T, Ihara Y, Takeda M. γ-secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 2013 Jan 31;3(1):42-51. PubMed.
Moore BD, Martin J, de Mena L, Sanchez J, Cruz PE, Ceballos-Diaz C, Ladd TB, Ran Y, Levites Y, Kukar TL, Kurian JJ, McKenna R, Koo EH, Borchelt DR, Janus C, Rincon-Limas D, Fernandez-Funez P, Golde TE. Short Aβ peptides attenuate Aβ42 toxicity in vivo. J Exp Med. 2018 Jan 2;215(1):283-301. Epub 2017 Dec 5 PubMed.
Bursavich MG, Harrison BA, Acharya R, Costa DE, Freeman EA, Hrdlicka LA, Jin H, Kapadnis S, Moffit JS, Murphy D, Nolan SJ, Patzke H, Tang C, Van Voorhies HE, Wen M, Koenig G, Blain JF, Burnett DA. Discovery of the Oxadiazine FRM-024: A Potent CNS-Penetrant Gamma Secretase Modulator. J Med Chem. 2021 Oct 14;64(19):14426-14447. Epub 2021 Sep 22 PubMed.
Ahn JE, Carrieri C, Dela Cruz F, Fullerton T, Hajos-Korcsok E, He P, Kantaridis C, Leurent C, Liu R, Mancuso J, Mendes da Costa L, Qiu R. Pharmacokinetic and Pharmacodynamic Effects of a γ-Secretase Modulator, PF-06648671, on CSF Amyloid-β Peptides in Randomized Phase I Studies. Clin Pharmacol Ther. 2020 Jan;107(1):211-220. Epub 2019 Sep 11 PubMed.
Massachusetts General Hospital
We agree that γ-secretase modulators (GSMs) are an excellent alternative for regulating Aβ levels in a hopefully safer manner than what has been experienced with γ-secretase and β-secretase inhibitors.
My close colleague Steve Wagner and I have been developing GSMs since 2001. Most recently, we developed a novel pyradizine-derived GSM (GSM-776890), which is expected to enter into Phase I clinical trials for AD later this year at UCSD and MGH. GSM-776890 lowers Aβ42 (IC50 = 5nM) and, to a lesser extent, Aβ40, while increasing levels of Aβ38 (IC50 = 29nM).
These studies have been carried out in academia at UCSD and MGH under the leadership of Steve Wagner and have been supported by the Cure Alzheimer's Fund, the NIH Neurotherapeutics Blueprint, and the NIA.
References:
Rynearson KD, Ponnusamy M, Prikhodko O, Xie Y, Zhang C, Nguyen P, Hug B, Sawa M, Becker A, Spencer B, Florio J, Mante M, Salehi B, Arias C, Galasko D, Head BP, Johnson G, Lin JH, Duddy SK, Rissman RA, Mobley WC, Thinakaran G, Tanzi RE, Wagner SL. Preclinical validation of a potent γ-secretase modulator for Alzheimer's disease prevention. J Exp Med. 2021 Apr 5;218(4) PubMed.
Rynearson KD, Buckle RN, Herr RJ, Mayhew NJ, Chen X, Paquette WD, Sakwa SA, Yang J, Barnes KD, Nguyen P, Mobley WC, Johnson G, Lin JH, Tanzi RE, Wagner SL. Design and synthesis of novel methoxypyridine-derived gamma-secretase modulators. Bioorg Med Chem. 2020 Nov 15;28(22):115734. Epub 2020 Sep 1 PubMed.
Asceneuron SA
The patient CSF biomarker data from Oskar Hansson’s group provide a compelling rationale for a resurrection of the γ-secretase modulator (GSM) approach for Alzheimer’s disease. As a note of caution, however, it is worth highlighting that, in clinical trials, not a single brain-penetrant NSAID or non-NSAID like GSM has ever been advanced beyond short-term Phase 1 studies.
Given the industry's investment into amyloid-β lowering drug development programs at the time, it can be assumed that strategic considerations played less of a role in preventing clinical progression of these molecules. Rather, this points to potential safety issues in longer-term preclinical toxicity studies. Hence, the key question remains whether GSM drug development has been hampered by off-target binding (GSMs tend to be characterized by high lipophilicity / logP), or whether there was still some on-target toxicity remaining.
Overall, armed with the tremendous knowledge about the drug target and mechanism of intra-membranous cleavage, a rational drug discovery approach could help answering these questions and support the discovery of new drugs for a genetically validated drug target. I would anticipate that future success will depend on new chemical scaffolds, and the systematic profiling of new GSMs against a larger number of γ-secretase substrates and the various isoforms of the enzyme.
McGill
This important study not only provides essential information about the clinical association between longer (Aβ42) and shorter amyloid-β peptides (Aβ38), but also underlines the necessity to better understand the mechanisms of amyloid peptide clearance.
Beyond the traditional CSF biomarker Aβ42, which is indicative of cerebral amyloid plaque formation, both Aβ42 and Aβ40 reflect changed γ-secretase processive activity, when the ratio of their soluble forms in body fluids is altered. The so-termed protective role of shorter peptides originates from the notion that effective processing of APP-CTFs by γ-secretase results in higher levels of shorter fragments at the expense of longer intermediates of the two product lines: Aβ48-45-42-38 and Aβ49-46-43-40 (Munter et al., 2007).
Notably, we discovered an additional enzymatic activity for BACE1, whereby within the “amyloidolytic” pathway (Fluhrer et al., 2003; Kirabali et al., 2019; Liebsch et al., 2019), the enzyme can degrade longer Aβ peptides, including plaque-forming Aβ42 and Aβ40, into the common, non-toxic intermediate Aβ34. In our 2019 study (Liebsch et al., 2019) we reported that Aβ34 is elevated in CSF from individuals with mild cognitive impairment in the Amsterdam Dementia Cohort who later progressed to dementia. These findings indicate that both γ- and β-secretases influence soluble amyloid Aβ42 and Aβ40 levels, the γ-secretase via its “processive” activity and BACE1 via its amyloidogenic and amyloidolytic activities.
Moreover, in “test-tube” experiments we found that Aβ38 was efficiently cleaved into Aβ34, when we determined the efficiencies of recombinant BACE1 to recognize longer Aβ species as substrates. Thus, higher Aβ38 levels could originate from (i) increased γ-secretase “processive” activity in the Aβ48-45-42-38 pathway and (ii) from decreased amyloidolytic activity exerted by BACE1. In conclusion, Aβ38 may reflect effective processing activity by γ-secretase but also stem from altered clearance activity of BACE1 when the step from Aβ38 to Aβ34 is impaired.
In addition, BACE1 activity is high in AD (Fukumoto et al., 2002; Holsinger et al., 2002; Yang et al., 2003) and under such conditions, Aβ38 should be efficiently converted into Aβ34. To link findings reported by Cullen et al. to potentially beneficial effects of γ-secretase modulators (GSMs), the characteristics of Aβ38 as an intermediate product of the processive pathway and as a precursor of Aβ34 in the amyloidolytic pathway should be taken into account. Thus, the measurement of Aβ34 in cohorts like BioFINDER, ADNI and others could be meaningful.
References:
Fluhrer R, Multhaup G, Schlicksupp A, Okochi M, Takeda M, Lammich S, Willem M, Westmeyer G, Bode W, Walter J, Haass C. Identification of a beta-secretase activity, which truncates amyloid beta-peptide after its presenilin-dependent generation. J Biol Chem. 2003 Feb 21;278(8):5531-8. PubMed.
Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002 Sep;59(9):1381-9. PubMed.
Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002 Jun;51(6):783-6. PubMed.
Kirabali T, Rigotti S, Siccoli A, Liebsch F, Shobo A, Hock C, Nitsch RM, Multhaup G, Kulic L. The amyloid-β degradation intermediate Aβ34 is pericyte-associated and reduced in brain capillaries of patients with Alzheimer's disease. Acta Neuropathol Commun. 2019 Dec 3;7(1):194. PubMed.
Liebsch F, Kulic L, Teunissen C, Shobo A, Ulku I, Engelschalt V, Hancock MA, van der Flier WM, Kunach P, Rosa-Neto P, Scheltens P, Poirier J, Saftig P, Bateman RJ, Breitner J, Hock C, Multhaup G. Aβ34 is a BACE1-derived degradation intermediate associated with amyloid clearance and Alzheimer's disease progression. Nat Commun. 2019 May 20;10(1):2240. PubMed.
Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. EMBO J. 2007 Mar 21;26(6):1702-12. PubMed.
Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, Li R, Shen Y. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003 Jan;9(1):3-4. PubMed.
Make a Comment
To make a comment you must login or register.