Do Sleeper Viruses Awaken in ALS?
Quick Links
A hibernating retrovirus, prodded from slumber, can provoke neurodegeneration, according to a paper in the September 30 Science Translational Medicine. Human endogenous retrovirus-K pumps out viral RNA and proteins in a subset of amyotrophic lateral sclerosis cases, according to senior author Avindra Nath and colleagues at the National Institute of Neurological Disorders and Stroke in Bethesda, Maryland. Moreover, rousing HERV-K in cultured cells killed neurons, and doing the same in mice sickened them in an ALS-like manner. “We are pretty comfortable in saying that the virus can cause motor neuron damage,” Nath said, cautioning, “What we haven’t shown is that it is a cause of ALS.” Additionally, there is no evidence that HERV-K could be transmitted from person to person like an infectious virus.
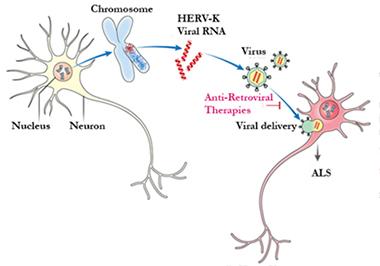
Insidious infection.
Once awakened in one neuron, HERV-K particles might induce neurodegeneration in its neighbors. If so, anti-retroviral therapy might help. [Courtesy of Avindra Nath.]
Nath studies a more famous retrovirus, the human immunodeficiency virus that gives rise to AIDS, and its neurological effects, such as dementia. In 2006, he treated a man who had both AIDS and an ALS-like syndrome, piquing his interest in the overlap between these two diseases. Once the patient started anti-retroviral therapy, his motor symptoms improved. As it turns out, scientists have reported a handful of such HIV cases over the years, and some regained their motor skills after anti-retroviral treatment. Scientists have also found reverse transcriptase, a marker for retrovirus activity, in the brain, serum, and cerebrospinal fluid of some people with sporadic ALS. However, they could not find evidence for infectious retroviruses associated with the disease, leading Nath to hypothesize that endogenous retroviruses might be involved (reviewed in Alfahad and Nath, 2013).
People possess scores of endogenous retroviral DNAs in their genomes, left over from retroviruses that copied themselves into human DNA at some point over the millennia and then fell dormant. These interloper sequences make up 8 to 10 percent of the human genome. In a 2011 paper, Nath and colleagues reported that DNA from one such intruder, HERV-K, was transcribed in the brain tissue of people who died of ALS, though they did not confirm it affected the disease process (see Dec 2010 news). The human genome houses dozens of copies of the HERV-K genome in various states of degradation compared with the active viral genome.
In the new study, first author Wenxue Li and colleagues followed up on that finding, asking if HERV-K contributes to neurodegeneration. Li examined brain tissue from 11 people who died of ALS and 16 controls. Nine of the cases had sporadic ALS, while one person carried a C9ORF72 expansion and another exhibited signs of both ALS and frontotemporal dementia. Li amplified RNA for HERV-K genes in those cases and controls. HERV-K expression varied from person to person, but overall was higher in ALS cases than in controls. Nath estimated that about 30 percent of the ALS cases possessed appreciable copy numbers of HERV-K RNA. The scientists found no correlation between clinical phenotypes and HERV-K expression levels.
The authors also stained postmortem tissue from 10 people who had died of ALS with antibodies for the HERV-K envelope protein and detected binding in cortical large pyramidal neurons and spinal cord anterior-horn neurons. HERV-K protein was absent from the brain tissue of 10 people who died of Alzheimer’s disease.
Could this reactivated, vintage virus do harm? Li transfected its genome, or just the envelope gene, into human neural cultures derived from pluripotent stem cells. The cells retracted their neurites and died. To discern whether triggering the endogenous HERV-K lurking in the genome could do the same, Li used a trick based on the CRISP/Cas9 gene editing system (see Sep 2014 series). Instead of altering the genome, he used the CRISPR/Cas9’s gene-homing ability to target transcription factors to an HERV-K gene. This turned the gene on, again causing the neurons to pull back projections and degenerate.
Next, co-author Myoung-Hwa Lee activated HERV-K in mice. As a quick test of its effects in animals, she electroporated the env gene into wild-type mouse brain before the pups were born. Two weeks after birth, the animals’ neurons appeared odd, with swollen beads along their neurites. Encouraged by this success, Lee generated a new line of transgenic mice, expressing the env gene under control of the Thy1 promoter in their neurons. They produced env RNA at levels about twice as high as seen in human brains. When Lee examined the tissues of 6-month-old mice, she observed their dendrites were shortened and less branched than normal, and the dendritic spines were fewer in number and abnormally shaped. Astrocytes ran amok, a common effect during neurodegeneration. As in human ALS, both upper and lower motor neurons degenerated. Compared with wild-type animals, the transgenic mice lacked about one-third of their corticospinal motor neurons and the volume of the motor cortex was reduced by about a fifth. Some parts of the spinal cord had barely any motor neurons.
These defects resulted in progressive motor symptoms. Compared with wild-type mice, the transgenics wandered less in an open field, and rested more often. They were quicker to tumble off a rotating rod, a common test of strength and coordination for ALS mice. Half were dead within 10 months. Defects were worse and progression faster than in the commonly used SOD1 model of ALS. “These env mice look like models that express genes associated with ALS,” said Josh Dubnau of Cold Spring Harbor Laboratory in New York.
Stanley Appel of the Houston Methodist Neurological Institute was not so sure. “I get the feeling that the pathology in this model is more in the brain than in the spinal cord,” he said, noting that lower-motor-neuron problems predominate in ALS. Neither Appel nor Dubnau were involved in the study.
The env expression was clearly toxic, but how? In the mice, the authors observed markers for double-stranded DNA breaks as well as disruption to the nucleolus. Nucleophosmin, normally a nucleolar membrane protein, had translocated to the cytoplasm. Via chromatin immunoprecipitation, Li found that HERV-K DNA bound directly to TDP-43, a protein implicated in ALS pathology. In the human neural cultures, Li determined that overexpression of TDP-43 activated HERV-K, while TDP-43 knockdown dampened its expression. In their previous study, TDP-43 expression correlated with that of the HERV-K reverse transcriptase, and the proteins co-localized, in the brain tissues of ALS cases. TDP-43 regulates transcription of numerous genes, including sequences in HIV and transposable elements, another kind of genomic stowaway (see Mar 2011 news; Nov 2010 news; Ou et al., 1995; Li et al., 2012). HERV-K expression depends on TDP-43, Nath concluded.
Dubnau noted that the idea of a retrovirus or retrotransposon-inducing chronic disease has precedent. For example, transposable Alu elements, when transcribed, induce a form of macular degeneration (Kaneko et al., 2011). Many cancer-causing viruses copy themselves into the host cell genome (Carrillo-Infante et al., 2007). HERV-W abets inflammation in multiple sclerosis, for which scientists are considering it as a treatment target or biomarker (Perron and Lang, 2010; Curtin et al., 2015). Herpes retrovirus for many years has been proposed as a pathogenic factor in sporadic Alzheimer’s, though the idea has not caught on (Harris and Harris, 2015; Feb 2011 webinar).
Dubnau speculated that HERV-K might be the first of many retroviruses or retrotransposons to be found stirring in ALS. He suggested it may be a “canary in the coal mine” signaling a general problem with silencing of toxic genetic elements. The DNA damage Li observed could result from the insertion of reverse-transcribed elements, he suggested. Dubnau and colleagues have discovered an endogenous retrovirus involved in neurodegeneration in fruit flies overexpressing TDP-43.
Scientists who spoke with Alzforum were enthusiastic about Nath’s study, and eager for more information about HERV-K and ALS. “It does explain why people found retroviral traces [in ALS] without any evidence of a real virus,” Appel said. “Nath's data points out elegantly that it is an activation of latent viral genomes that sit in all of us.” He was curious about whether HERV-K also awakens in genetic cases of ALS, particularly TDP-43 cases. Jeremy Garson of University College London, who did not participate in the research, noted that it raises numerous questions about what subsets of ALS might express HERV-K, whether people with the disease have extra copies of the integrated viral genome or activate certain variants, and what other factors might spur HERV-K revival. “It will be important for the major findings of this work to be replicated independently by other research groups,” he added (see full comment below).
Robert Brown of the University of Massachusetts Medical School in Worcester and Ammar Al-Chalabi of King’s College London, who co-wrote an editorial commentary in the same issue of Science Translational Medicine, agree the study raises many new questions. “Perhaps most fundamental is the proverbial chicken-and-egg challenge: Is the activation of HERV-K a cause or consequence of motor neuron degeneration?” they wrote. In addition, “Can one now define a cocktail of anti-retroviral interventions that are beneficial, and will the activity of the retrovirus prove to be a quantitative biomarker of disease activity?”
Nath is working on answering these questions, as well as the possibility of treating retroviral activation in ALS. He recently secured approval for a Phase 1 trial of a four-part anti-retroviral cocktail. For now, he aims not to necessarily treat the disease, but just to find out if the medications curb HERV-K titers in the blood. Nath cautioned that people with ALS should not try this treatment outside of a trial. “These are not innocuous drugs, and we do not know if the cocktail will work,” he pointed out. Typically, such treatments affect disease course only if they almost completely eliminate the retrovirus, he added.
Dubnau wondered if any HERV-K retroviral DNA hiding in the human genome can produce a competent, infectious virus. If so, it might transmit ALS from cell to cell (see image above). Appel believes the viral spread would likely incorporate glia somehow, as well as neurons. Scientists have labelled HERV-K a “dead” virus, able to express toxic gene products but not assemble a complete infectious particle. However, they based that conclusion on the analysis of just a few reference DNA samples, which indicated the HERV-K sequences in the human genome were incomplete or contained point mutations. Might some people have a functional HERV-K genome, waiting to be revived? To determine that will be difficult, Dubnau noted, because there are many copies of repetitive sequences that are challenging to assemble by short-read sequencing. Nonetheless, Nath has started a project to examine HERV-K copy number and polymorphisms. He also plans to look for cell-to-cell spread of HERV-K in vitro, and research possible interventions in his animal model. Furthermore, Nath plans to explore HERV-K in tissue samples from people who had frontotemporal dementia, which includes TDP-43 proteinopathy in some cases.—Amber Dance
References
News Citations
- Does an Ancient Retrovirus Come Out of Hiding in ALS?
- CLIPs of TDP-43 Provide a Glimpse Into Pathology
- San Diego: TDP-43 Targets Loom Large—But Where’s the Bull’s Eye?
Series Citations
Webinar Citations
Paper Citations
- Alfahad T, Nath A. Retroviruses and amyotrophic lateral sclerosis. Antiviral Res. 2013 Aug;99(2):180-7. PubMed.
- Ou SH, Wu F, Harrich D, García-Martínez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995 Jun;69(6):3584-96. PubMed.
- Li W, Jin Y, Prazak L, Hammell M, Dubnau J. Transposable elements in TDP-43-mediated neurodegenerative disorders. PLoS One. 2012;7(9):e44099. Epub 2012 Sep 5 PubMed.
- Kaneko H, Dridi S, Tarallo V, Gelfand BD, Fowler BJ, Cho WG, Kleinman ME, Ponicsan SL, Hauswirth WW, Chiodo VA, Karikó K, Yoo JW, Lee DK, Hadziahmetovic M, Song Y, Misra S, Chaudhuri G, Buaas FW, Braun RE, Hinton DR, Zhang Q, Grossniklaus HE, Provis JM, Madigan MC, Milam AH, Justice NL, Albuquerque RJ, Blandford AD, Bogdanovich S, Hirano Y, Witta J, Fuchs E, Littman DR, Ambati BK, Rudin CM, Chong MM, Provost P, Kugel JF, Goodrich JA, Dunaief JL, Baffi JZ, Ambati J. DICER1 deficit induces Alu RNA toxicity in age-related macular degeneration. Nature. 2011 Mar 17;471(7338):325-30. Epub 2011 Feb 6 PubMed.
- Carrillo-Infante C, Abbadessa G, Bagella L, Giordano A. Viral infections as a cause of cancer (review). Int J Oncol. 2007 Jun;30(6):1521-8. PubMed.
- Perron H, Lang A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin Rev Allergy Immunol. 2010 Aug;39(1):51-61. PubMed.
- Curtin F, Perron H, Faucard R, Porchet H, Lang AB. Treatment against human endogenous retrovirus: a possible personalized medicine approach for multiple sclerosis. Mol Diagn Ther. 2015 Oct;19(5):255-65. PubMed.
- Harris SA, Harris EA. Herpes Simplex Virus Type 1 and Other Pathogens are Key Causative Factors in Sporadic Alzheimer's Disease. J Alzheimers Dis. 2015;48(2):319-53. PubMed.
External Citations
Further Reading
Papers
- Ravits J. Sporadic amyotrophic lateral sclerosis: a hypothesis of persistent (non-lytic) enteroviral infection. Amyotroph Lateral Scler Other Motor Neuron Disord. 2005 Jun;6(2):77-87. PubMed.
- De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R, Garaci E, Grassi C, Palamara AT. Infectious Agents and Neurodegeneration. Mol Neurobiol. 2012 Dec;46(3):614-638. PubMed.
Primary Papers
- Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, Maric D, Morris HD, Lentz M, Pak K, Mammen A, Ostrow L, Rothstein J, Nath A. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med. 2015 Sep 30;7(307):307ra153. PubMed.
- Brown RH Jr, Al-Chalabi A. Endogenous retroviruses in ALS: A reawakening?. Sci Transl Med. 2015 Sep 30;7(307):307fs40. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
Several prior strands of evidence (mainly indirect) pointed to the possibility of retroviral involvement in ALS. These can be summarized as follows:
The new study expands on earlier work by the same authors (Jeffrey Rothstein and Avindra Nath), which suggested, on the basis of preliminary findings, that the source of the reverse transcriptase in ALS patients may be the human endogenous retrovirus HERV-K (Douville et al., 2011). The new study is substantial and impressive in that it has employed a wide range of powerful in vivo and in vitro molecular biological, immunological, genetic, and other techniques to demonstrate that (a) HERV-K is expressed in the brain of ALS patients at higher levels than in controls, (b) HERV-K expression in human neurons causes toxicity, (c) HERV-K expression in transgenic mice causes a form of motor neuron disease with some similarities to ALS, and (d) HERV-K expression may be regulated by the DNA-binding protein, TDP-43.
A key question is whether the increased expression of HERV-K in the brains of ALS patients is a cause or a consequence of the disease. In other words, is it an essential part of the pathogenic process or simply an epiphenomenon? In their new study, Li and colleagues have gone some way to answering this by demonstrating not only that elevated HERV-K expression can cause neurotoxicity but also that the elevated expression is not just a non-specific consequence of neuronal injury. The new data presented in the paper represent a significant contribution to our understanding of the potential role of endogenous retroviruses in neurological disease.
In general, the methodology employed seems appropriate, although there are a few minor concerns. First, the RT-PCR assay used to measure HERV-K RNA expression in brain and neuronal cultures employed GAPDH RNA as a reference for normalization without demonstrating that GAPDH expression levels were stable. For accurate measurement of HERV-K RNA by this relative quantification method it is essential to know that the GAPDH RNA levels were the same in the ALS and control brains. The importance of validating reference genes in RT-PCR is emphasized in the MIQE Guidelines (Bustin et al., 2009) as follows: “Normalization against a single reference gene is not acceptable unless the investigators present clear evidence for the reviewers that confirms its invariant expression under the experimental conditions described.” Ideally, full MIQE (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) details should have been included within the supplementary materials.
Second, the demonstration of HERV-K RNA expression in HERV-K transfected human neurons in culture was not accompanied by proof of concomitant protein expression.
Third, the attempt to demonstrate the specificity of HERV-K elevated expression in ALS by measuring the expression of other HERV families was not very comprehensive, and importantly failed to measure HERV-W expression. HERV-W expression levels would have been of particular interest here because HERV-W has been implicated in other human neurological/psychiatric disorders, including multiple sclerosis and schizophrenia.
It will be important for the major findings of this work to be replicated independently by other research groups. In addition, future questions to be addressed should include the following:
The regulation of endogenous retroviral latency is complex and a variety of agents, including physical, chemical, infectious, and immunologic, have been shown to be able to induce their activation. What infectious or other environmental factors might induce abnormal expression of HERV-K in ALS patients?
The work described in this paper is unlikely to have immediate practical implications for patients currently suffering from ALS, although it is quite possible that the findings may initiate the search for novel approaches to therapy, directly or indirectly targeting retroviral nucleic acids, proteins, or associated pathways. It may be appropriate to consider establishing formal clinical trials of existing anti-retroviral drugs.
For researchers in the field, these findings add further support for the theory of retroviral involvement in ALS. The HERV-K transgenic mouse may have potential as an animal model to facilitate the development and testing of existing and novel therapeutic agents for the treatment of patients with ALS.
References:
McCormick AL, Brown RH Jr, Cudkowicz ME, Al-Chalabi A, Garson JA. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology. 2008 Jan 22;70(4):278-83. PubMed.
Steele AJ, Al-Chalabi A, Ferrante K, Cudkowicz ME, Brown RH Jr, Garson JA. Detection of serum reverse transcriptase activity in patients with ALS and unaffected blood relatives. Neurology. 2005 Feb 8;64(3):454-8. PubMed.
Andrews WD, Tuke PW, Al-Chalabi A, Gaudin P, Ijaz S, Parton MJ, Garson JA. Detection of reverse transcriptase activity in the serum of patients with motor neurone disease. J Med Virol. 2000 Aug;61(4):527-32. PubMed.
Douville R, Liu J, Rothstein J, Nath A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol. 2011 Jan;69(1):141-51. PubMed.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009 Apr;55(4):611-22. Epub 2009 Feb 26 PubMed.
University of Chicago
This interesting and provocative paper carries on a theme that has been pursued for a number of years by Nath and colleagues. The investigators find that a subset of ALS patients has increased expression of HERV-K genes and increased immunoreactivity compared to controls of the HERV-K envelope (env) protein in motor neurons in autopsy tissue. In vitro studies show that overexpression of env leads to degeneration of IPSC-derived human neurons. Furthermore, transgenic mice that overexpress env under the thy-1 promoter have motor deficits, loss of motor neurons, denervation of muscle, and early death. The investigators show that TDP-43, which is mislocalized in most cases of ALS, binds to the HERV-K LTR, thereby leading to transactivation of HERV-K.
It is instructive to review motor neuron diseases associated with mouse retroviruses. Infection of mice with a temperature-sensitive mutant of the Moloney murine leukemia virus, called ts1, causes progressive non-inflammatory hind-limb paralysis that is thought to result from apoptosis of glial cells leading to ER stress in neurons and culminating in spongiform changes in the brain stem and ventral horn of the spinal cord (Kim et al., 2004); transgenic mouse studies indicated that expression of env led to disease. Another related murine leukemia virus, Cas-Br-E-MuLV, also produces paralysis with similar spongiform changes concentrated in the lumbar spinal cord anterior horn (Kay et al., 1993). As with ts1, infection of non-neuronal cells and a key role of env (as suggested by chimeric virus and transgenic mouse studies) have been implicated in disease pathogenesis. More complicated is age-dependent poliomyelitis, a murine disease that requires multiple proviral copies of an endogenous murine leukemia virus, infection with lactic dehydrogenase virus, a permissive allele in the mouse, and an immunocompromised state (Contag et al., 1992). Of note, the immunocompromised state could result from age, which, interestingly, is the most significant risk factor in developing sporadic ALS.
Several questions arise from the paper by Nath and colleagues. The paper’s focus on the effect of env makes one wonder about the effect of gag and pol, the two other major genes encoded by retroviral genomes. How efficiently is HERV-K transactivated when TDP-43 is mislocalized to the cytoplasm, as is true in most cases of ALS? Do cases of mutant SOD1-induced familial ALS have increased expression of HERV-K despite the reported absence of TDP-43 abnormalities? Is HERV-K expression increased in IPSC-derived motor neurons from sporadic cases of ALS or from familial ALS patients with mutations of C9ORF72?
The authors are appropriately cautious about the role of HERV-K in ALS pathogenesis since the mislocalization of TDP-43 causes varied splicing abnormalities with abnormal expression of multiple genes, including, as demonstrated in the present paper, those of an endogenous retrovirus. It is likely that there are a number of bullets that contribute to the death of motor neurons in ALS.
References:
Kay DG, Gravel C, Pothier F, Laperrière A, Robitaille Y, Jolicoeur P. Neurological disease induced in transgenic mice expressing the env gene of the Cas-Br-E murine retrovirus. Proc Natl Acad Sci U S A. 1993 May 15;90(10):4538-42. PubMed.
Kim HT, Waters K, Stoica G, Qiang W, Liu N, Scofield VL, Wong PK. Activation of endoplasmic reticulum stress signaling pathway is associated with neuronal degeneration in MoMuLV-ts1-induced spongiform encephalomyelopathy. Lab Invest. 2004 Jul;84(7):816-27. PubMed.
Contag CH, Harty JT, Plagemann PG. Pathogenesis of age-dependent poliomyelitis of mice. In: Molecular Neurovirology Pathogenesis of Viral CNS Infections. Ed. R.P. Roos. Humana Press, 1992.
University of Sassari
This paper by Li and colleagues on the contribution of endogenous retroviruses (HERV) of the K family to the pathogenesis of ALS is exciting. Currently, ALS is a disease without effective treatments. Therefore, this insight into its possible pathogenesis opens new horizons and, hopefully, new room to manoeuvre.
For more than 16 years I have studied another HERV family member, HERV-W, in the onset and neuro-pathogenesis of multiple sclerosis (MS). I knew of the existence of HERV-K expression in ALS from a poster by the Dr. Nath’s group at the 2013 meeting of the International Society of Neurovirology. What made me literally jump then was the sight of a nice, clear, electrophoretic band of what looked as a retro-transcribed cDNA from a full-length HERV genome, extracted from a brain sample of an ALS patient. Since then, every time I met Dr. Nath, I expressed my deep interest about their findings and the role of HERV-K in ALS neurodegeneration
MS and ALS are quite different: Primarily, ALS affects neurons in the gray matter of the brain, while MS affects oligodendrocytes in the white matter. The behavior and progression of the two diseases have completely different patterns. However, both diseases start focally, and then the pathology spreads. The etiology of both diseases is uncertain, complex, and multifactorial. A genetic component is postulated for MS and, at least partly, for ALS, while some interaction(s) between genetic and environmental factors are believed to influence both.
For MS, several independent studies, including ours, have shown a direct correlation between disease onset/ progression and expression of HERV-W/MSRV/Syncytin-1. The HERV-Wenv protein has neuropathogenic and immunopathogenic properties, both in vitro and in animal models, that can account for MS neurodegeneration. HERV-Wenv protein has been detected in glial cells surrounding the degenerating nerve sheaths. Currently, a Phase 2 clinical trial of an antibody to the HERV-Wenv protein is in progress for MS patients and seems promising.
In the ALS cases studied by Li and colleagues, the HERV-Kenv protein is detected within cells relevant to the ALS disease and it is activated by the TDP-43 protein, a hallmark of ALS. Moreover, transgenic mice expressing the HERV-Kenv protein developed progressive motor dysfunctions, reminiscent of human ALS.
It is not surprising that HERV-Kenv has these effects. The env proteins that make up the external envelope, or pericapsid, of retroviruses, including the gp120 env of the HIV virus and the aforementioned HERV-Wenv, have several neuropathogenic properties.
In my opinion, the reason why this finding of HERV-K in an ALS subset is important is that it raises the possibility, so far for some patients only, of changing ALS by targeting the virus.
If further studies confirm HERV-Kenv expression in other ALS cohorts, and if its levels turn out to correlate with disease progression, then we can be allowed to hope that future innovative treatments aimed at HERV-K inhibition may attenuate ALS pathology.
I am acutely aware that such optimism may be easily dashed, but this is a research path that deserves deeper consideration.
Victoria University
Even though I like the study, which is solid and agreeable in part, I have some philosophical points to make that may help unveil more concrete associations between ERV and human disease. I believe that it is too early to conclude that HERV-K expression within neurons of patients with ALS contributes to neurodegeneration and disease pathogenesis. This is because most HERVs are harmless and all human cells and all individuals have essentially the same set of HERVs. Thus, the negative correlations shown by Nath’s group show only indirectly how an HERV can be tied to a disease in humans—in this case ALS. The proof that HERV is the etiological agent of ALS may never materialize, as the Koch postulates or similar rules adapted to present-day techniques mainly pertain only to exogenous agents.
Eight percent of the human genome consists of human endogenous retroviruses, or HERVs, and, if we extend this to HERV fragments and derivatives, the retroviral legacy amounts to roughly half our DNA (Ryan, 2004). Interestingly, both human class I and class II MHC genes carry a high density of HERV elements compared to other multi-locus-gene families (Bartholomew and Ihle, 1991). Striking to note is that HERVs have contributed to the formation of extensively duplicated duplicon blocks that make up the HLA class I family of genes inherited as immuno-haplotypes (Bartholomew and Ihle, 1991). In this context, it is important to emphasize that the majority of ERVs found in vertebrate genomes are of ancient nature, inactivated by mutation, and have reached genetic fixation in the host species they have evolved in. Therefore, they are extremely unlikely to have negative effects on their hosts except under very unusual circumstances.
Although not applicable to the present study, where only ALS patients were tested, it is important to note that Nath’s group had previously reported HIV-positive ALS patients testing positive for HERV antibodies. In this context it is important to iterate that viruses can interfere with the same pathways and anatomical structures in the brain that are involved in neurologic diseases, such as Alzheimer’s, Parkinson’s, ALS, and other dementias. Support for this argument comes from antiretroviral agents reverting dementia in HIV patients (Zhou et al., 2013). Thus, taking this and the previous study, the striking observations made by Nath’s group warrant a detailed investigation into the possible genetic interactions between HERV and other retroviruses and their role in neurodegeneration as a consequence of their weakening the immune system due to age.
We have learned a lesson from an evolutionarily conserved ERVWE1 or "syncytin" gene—a derivative of ERV insertion that is vital to the creation of human progeny. Its tight conservation across primate species in the same genomic locus speaks volumes for its functional significance. As we understand now that microbes are integral to our very survival by guiding and managing our immune system against foreign attack, it is time to rethink how the genomic remnants of microbial elements, with which we have co-evolved, interact with other exogenous agents in vivo.
Although the study by Nath’s group is exciting and the investigators were cautious not to over-interpret that HERVs cause ALS, it is important to consider the evolutionary and philosophical parts of the debate on ERVs. Extrapolating from in vitro findings may not be meaningful simply because they may not jibe when compared with the in vivo functionality of ERV elements. The inheritance of ERV elements between hosts and their genetic and functional conservation in humans and mammals are some of the points that can shed more light on which direction to pursue to determine whether ERVS are pathogenic.
References:
Ryan FP. Human endogenous retroviruses in health and disease: a symbiotic perspective. J R Soc Med. 2004 Dec;97(12):560-5. PubMed.
Bartholomew C, Ihle JN. Retroviral insertions 90 kilobases proximal to the Evi-1 myeloid transforming gene activate transcription from the normal promoter. Mol Cell Biol. 1991 Apr;11(4):1820-8. PubMed.
Zhou L, Miranda-Saksena M, Saksena NK. Viruses and neurodegeneration. Virol J. 2013 May 31;10:172. PubMed.
Make a Comment
To make a comment you must login or register.