Do Perivascular Macrophages Mediate Aβ Pathology?
Quick Links
Amyloid-β is bad news for neurons, and also for the blood vessels that feed them. Now, a study from the lab of Costantino Iadecola, Weill Cornell Medical College in New York, proposes an unexpected role for perivascular macrophages (PVMs) as mediators of Aβ-induced vascular dysfunction in mice. First author Laibaik Park and colleagues propose that PVMs respond to Aβ by releasing a burst of reactive oxygen species (ROS) that put the brakes on blood flow. The findings appeared in the May 17 Circulation Research.
“Park and colleagues have put together an elegant study that links activation of perivascular macrophages (PVMs) with known effects of Aβ on vascular function,” wrote Berislav Zlokovic and colleagues at the University of Southern California, Los Angeles, in an email to Alzforum. “This is an exciting discovery, which identifies a new target for developing treatments for the vascular dysfunction that often accompanies AD and other diseases,” they wrote. (See complete comment below.)
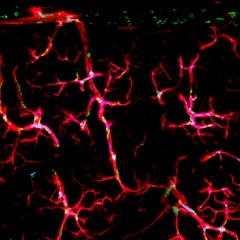
Perivascular macrophages (green) juxtaposed to arterioles (red) in the somatosensory cortex of Tg2576 mice produce reactive oxygen species appear (blue). [Courtesy of Costantino Iadecola and Labaik Park.]
Previously, Iadecola had shown that Aβ profoundly affects the coupling between neuronal activity and blood flow in mice. Compared with wild-type mice, Tg2576 animals, which overexpress amyloid precursor protein, have both lower resting cerebral blood flow and diminished capacity to increase that flow in response to synaptic activity—a property known as neurovascular coupling. Iadecola’s earlier work established that Aβ reduces this coupling through a pathway that involves the innate immunity receptor CD36, NADPH oxidase, the production of ROS, and damage to endothelial cells that regulate blood flow (Mar 2011 news). However, the identity of the ROS-producing cells was a mystery.
Perivascular macrophages seemed like good candidates. A unique population of brain-resident phagocytic cells derived from bone marrow, PVMs are perfectly positioned to encounter Aβ as it collects and is cleared through the Virchow-Robin space surrounding cerebral blood vessels. PVMs express both CD36 and NADPH oxidase, and have the capacity to make large quantities of ROS. To investigate the function of PVMs, Iadecola’s group transiently ablated the cells using liposomal clodronate, a cell toxin that is selectively taken up by phagocytizing cells. In their hands, intraventrical administration of the liposomal preparation resulted in an 80 percent decrease in phagocytic cells in the brain, including PVMs and meningeal macrophages. They found no detectable effect on the numbers of microglia, smooth muscle cells, astrocytes, or endothelial cells.
Unlike in intact animals, in mice depleted of PVMs, Aβ applied directly to the cortex did not interfere with resting cerebral blood flow, or with somatosensory cortex-neurovascular coupling evoked by stroking a whisker. Production of ROS in response to Aβ was also much lower in PVM-depleted mice. Similarly, in young (three- to four-month-old) Tg2576 mice, ablation of PVMs normalized cerebral blood flow and activity-coupled flow, and reduced oxidative stress.
To confirm that CD36- and NADPH oxidase-expressing cells mediated these responses, the investigators used a bone marrow transplant model. After eliminating native PVMs by lethal irradiation, they reconstituted the mice with donor marrow from wild-type animals, or animals lacking CD36 or the Nox2 subunit of NADPH oxidase. In mice transplanted with either knockout, topical Aβ now elicited no neurovascular dysfunction or ROS production. Likewise, in irradiated Tg2576 mice, transplantation of CD36 or Nox2 knockout bone marrow normalized blood flow and ROS levels. In a converse reconstitution, they showed that transplantation of wild-type bone marrow could restore Aβ responsiveness to CD36 knockout mice.
The unique combination of ablation, bone marrow chimeras, knockouts, and Tg2576 AD model makes a strong case for a role of perivascular macrophages in the response to Aβ. Together, this work highlights a role for the immune system, said Donna Wilcock of the University of Kentucky in Lexington. “Finding that mediator opens up a whole new line of research to study how AD pathology interacts with and affects the vasculature,” she told Alzforum. For example, Wilcock wondered about immunotherapies, which cause the clearance of Aβ through the Virchow-Robin space. “Could it be that Aβ ends up finding these perivascular macrophages and disrupting neurovascular coupling?” she asked.
Wilcock would like to know more about what forms of Aβ trigger the PVMs. The young mice used in the current study would have mainly soluble Aβ, and it is unclear how the results might change with fibrillar forms, or in the presence of plaques that are seen in older mice and people, she said.
Since they used young mice, the authors do not know if ablation of PVMs, and the better circulation that affords, improves cognition. The Tg2576 mice they used for the ablation and bone marrow experiments would not have developed memory problems yet. Iadecola told Alzforum his group is now doing experiments in older animals, to see if reversing blood flow abnormalities can improve memory.—Pat McCaffrey
References
Research Models Citations
News Citations
Further Reading
Primary Papers
- Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, Younkin L, Younkin S, Zhou P, Carlson G, Anrather J, Iadecola C. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ Res. 2017 Jul 21;121(3):258-269. Epub 2017 May 17 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of South Alabama
University of Southern California, Keck School of Medicine
University of Southern California
Park and colleagues have put together an elegant study that links activation of perivascular macrophages (PVMs) with known effects of Aβ on vascular function. Importantly, they have identified PVMs as cells responsible for a significant portion of reactive oxygen species (ROS) generated by Aβ exposure, and the mechanism is CD36- and Nox2-dependent. ROS are known to inhibit vascular reactivity in mouse models of beta amyloidosis, but it was previously thought that a considerable portion of ROS generated in the vascular periphery came from endothelial cells. This is an exciting discovery that identifies a new target for developing treatments for vascular dysfunction that often accompanies AD and other diseases. Furthermore, the necessity of CD36 in this process begs the question of how innate immunity may play a role in neurovascular dysfunction.
Karolinska Institute
Perivascular macrophages (PVM) in the Virchow-Robin space around blood vessels in the brain parenchyma have been implicated for a long time in the pathology of brain diseases, including Alzheimer's disease (AD). This recent study demonstrates very elegantly that PVMs are an important source of reactive oxygen species (ROS) and that Aβ increases their ROS production. Aβ is also shown to impair neurovascular function by reducing the resting cerebral blood flow and by diminishing the blood flow increase normally observed in the somatosensory cortex following whisker stimulation. By manipulating the number of PVMs or their capacity to produce ROS, the authors link ROS production of PVM to the Aβ-induced neurovascular dysfunction. Three very different ways were used to increase Aβ levels in the perivascular space of living mice: the cortex was either superfused with Aβ during the experiment; increased levels of human mutated Aβ were endogenously produced in the brain parenchyma of the transgenic AD mouse model Tg2576; or Aβ was injected into the blood flow of wild-type mice and reached the perivascular space through endothelial cells. In all three experimental setups, Aβ induced similar neurovascular dysfunction, and depletion of PVMs or their ROS production ameliorated the Aβ effect.
These results are compelling and point to new links between cells of the innate immune system, Aβ, and neurovascular dysfunction. This leads to important questions. Does it matter for the pathophysiology of AD? Is the effect strong enough to impact on brain function? Is the mechanism important under the conditions of the human (aging) brain and human immune system? The authors are convinced that, based on their results, it is reasonable to expect that PMV depletion will improve cognition.
In my opinion it will not be an easy task to determine if this mechanism plays an essential role in the pathophysiology of AD.
It is puzzling that Aβ effects on PVM and neurovascular function have been demonstrated in three- to four-month-old Tg2576 mice long before robust cognitive deficits are described in this mouse line (six months and older). This suggests that the change of cerebral blood flow in response to neuronal activity, as demonstrated for the whisker stimulation in the somatosensory cortex, is not important for cognitive performance. How do neurons cope so well for several months if Aβ is impeding the increase in blood flow-inactive cortical areas?
The authors report briefly on two failed attempts to experimentally prove that abolishing ROS production in PVM or depleting PVM altogether improves cognition in the Tg2576 mice. One suggestion would be to prolong the cortical PVM depletion by repeated intraventricular injections of clodronate. This would expose animals to multiple surgical procedures, which impacts heavily on behavioral tests, but it might help prove the point.
Tg2576 mice are one of the best-characterized and frequently used AD mouse models. Nevertheless, it would be helpful to include other AD mouse models, with more physiological levels of Aβ expression, into future studies. In the Tg2576 mice the APP expression is driven by the hamster prion promoter, leading to 14-fold elevated Aβ levels. Using other mouse lines could rule out that the described Aβ effect is triggered only at very high Aβ levels.
Furthermore, it is important to point to limitations of mouse models of AD, which reproduce only a few features of the complex brain disease, such as the elevated or mutant APP expression, plaque formation, dendritic spine loss, or some cognitive impairments. Other pathological changes involving the immune system and interactions between immune and nervous system are more difficult to reproduce and study in animal models. For example, more immune cells, including macrophages, have been reported in human aging brain-parenchyma than in mouse, in particular in pathological conditions such as AD. Most rodent AD models do not reproduce these features.
However, attempts to reduce specifically the number of PVMs in the human brain for long periods of time in order to improve cognition might be not only challenging but also dangerous, considering their important role in the innate immune defense. Compared to the almost sterile conditions mice face in research animal facilities, we humans live in a very complex world and are exposed to many pathogens.
Last, but not least, I would like to emphasize that the idea that reducing ROS levels in the perivascular space in the brain will improve cognition in any stage of AD is simplistic and fallacious. ROS have multiple roles, including as signaling molecules and defense mechanisms, but that is surely old news by now.
Weill College Medicine
Weill Cornell Medical College
We thank Drs. Nelson, Kisler, and Zlokovic for their positive comments on our paper.
The role of innate immunity receptors in AD is an emerging area of great interest, fostered by GWAS and whole-genome sequencing studies that have suggested their association to the disease. In this context, RAGE (receptors for advanced glycation end-products), studied extensively by Dr. Zlokovic and colleagues, are expressed in macrophages and their investigation may provide further insight into this PVM-based mechanism. This point is particularly relevant since CD36 may form macromolecular complexes with RAGE and act though cooperative signaling.
Weill College Medicine
Weill Cornell Medical College
We thank Dr. Galter for the careful read of our paper and the interest in this work. We agree that demonstration of functional (cognitive) rescue is a necessary step to assess the translational relevance of the cerebrovascular rescue, and that APP overexpression in Tg2576 mice is an incomplete recapitulation of AD.
The fact that in presymptomatic Tg2576 mice, as shown in presymptomatic individuals with AD (Ruitenberg et al., 2005; Iturria-Medina et al., 2016; Wierenga et al., 2014; Okonkwo et al., 2014), reviewed in Kisler et al., 2017, the vascular dysfunction is not associated with cognitive deficits and is consistent with the need for the dysfunction to be prolonged and work in concert with emerging Aβ (in Tg2576 mice) and tau pathology (in AD). The nitric oxide deficit associated with neurovascular dysfunction may also induce tau pathology (Austin and Katusic, 2016) and suppress hippocampal synaptic plasticity (Hopper and Garthwaite, 2006), providing diverse paths to neuronal dysfunction beyond the reduction in blood flow. However, we need to learn more about the effect of neurovascular dysfunction on neuronal circuits involved in cognition.
Is the neurovascular effect strong enough to improve cognition? We concur that is hard to predict based on the available evidence, but we are encouraged by the following observations.
As for the potential deleterious effects of radical depletion, this point is well taken since radicals may be signaling molecules in the vasculature. To minimize this concern, the idea would be to selectively disrupt a specific enzymatic source of ROS (NOX2) in a specific cell type, the perivascular macrophage, in which ROS production is well known to induce oxidative stress and cellular dysfunction and damage. But we agree with Dr. Galter that, as for all immunomodulation-based therapies, the possibility of infectious or immunological complications cannot be ruled out.
We would like to take this opportunity to thank Dr. Galter again for her insightful and constructive appraisal of our work.
References:
Ruitenberg A, den Heijer T, Bakker SL, van Swieten JC, Koudstaal PJ, Hofman A, Breteler MM. Cerebral hypoperfusion and clinical onset of dementia: the Rotterdam Study. Ann Neurol. 2005 Jun;57(6):789-94. PubMed.
Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative. Early role of vascular dysregulation on late-onset Alzheimer's disease based on multifactorial data-driven analysis. Nat Commun. 2016 Jun 21;7:11934. PubMed.
Wierenga CE, Hays CC, Zlatar ZZ. Cerebral blood flow measured by arterial spin labeling MRI as a preclinical marker of Alzheimer's disease. J Alzheimers Dis. 2014;42 Suppl 4:S411-9. PubMed.
Okonkwo OC, Xu G, Oh JM, Dowling NM, Carlsson CM, Gallagher CL, Birdsill AC, Palotti M, Wharton W, Hermann BP, LaRue A, Bendlin BB, Rowley HA, Asthana S, Sager MA, Johnson SC. Cerebral Blood Flow is Diminished in Asymptomatic Middle-Aged Adults with Maternal History of Alzheimer's Disease. Cereb Cortex. 2012 Dec 12; PubMed.
Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017 Jul;18(7):419-434. Epub 2017 May 18 PubMed.
Austin SA, Katusic ZS. Loss of Endothelial Nitric Oxide Synthase Promotes p25 Generation and Tau Phosphorylation in a Murine Model of Alzheimer's Disease. Circ Res. 2016 Oct 28;119(10):1128-1134. Epub 2016 Sep 6 PubMed.
Hopper RA, Garthwaite J. Tonic and phasic nitric oxide signals in hippocampal long-term potentiation. J Neurosci. 2006 Nov 8;26(45):11513-21. PubMed.
Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A. 2008 Jan 29;105(4):1347-52. PubMed.
Park L, Zhou J, Zhou P, Pistick R, El Jamal S, Younkin L, Pierce J, Arreguin A, Anrather J, Younkin SG, Carlson GA, McEwen BS, Iadecola C. Innate immunity receptor CD36 promotes cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2013 Feb 19;110(8):3089-94. PubMed.
Faraco G, Sugiyama Y, Lane D, Garcia-Bonilla L, Chang H, Santisteban MM, Racchumi G, Murphy M, Van Rooijen N, Anrather J, Iadecola C. Perivascular macrophages mediate the neurovascular and cognitive dysfunction associated with hypertension. J Clin Invest. 2016 Dec 1;126(12):4674-4689. Epub 2016 Nov 14 PubMed.
Make a Comment
To make a comment you must login or register.