In DIAN, Serial and Cross-Sectional Biomarker Trajectories Diverge
Quick Links
Kiss those smooth, sigmoidal curves goodbye, and embrace the jumbled complexity of spaghetti plots. Longitudinal data charting the trajectories of a bevy of biomarkers in people with autosomal-dominant AD (ADAD) was published September 20 in Neurology. For some biomarkers, the serial measurements confirmed previous cross-sectional data. However, for cerebrospinal fluid tau, serial changes told a different story. Notably, the longitudinal data indicated that phospho-tau levels plummeted five years prior to symptom onset. Cross-sectional data has had CSF p-tau climb during that period. Led by Randall Bateman at Washington University in St. Louis, the study may have implications for how the biomarkers are used in clinical trials.
- In DIAN, serial data tell slightly different story than cross-sectional.
- CSF tau starts high, then reaches a plateau; p-tau rises, then falls prior to onset.
- P-tau plummet could signify spread of tau tangles in brain.
Henrik Zetterberg of the University of Gothenburg in Sweden said having repeated measurements in the same individuals was powerful, and pointed to underlying biological processes reflected by the biomarkers. In particular, he believes the dramatic drop in CSF phospho-tau happens when neurofibrillary tangles start spreading and sequestering tau.
The Dominantly Inherited Alzheimer’s Network (DIAN) started recruiting members of families carrying autosomal-dominant mutations in APP, PSEN1, and PSEN2 in 2009. Ever since, researchers have steadily tracked cognition and multiple biomarkers in the cohort, which includes noncarriers as well as symptomatic and asymptomatic carriers. While longitudinal data was dribbling in, researchers had analyzed multiple cross-sectional studies to estimate how markers changed over time (Jul 2010 news; Jul 2012 news; Jun 2013 news). Through these cross-sectional studies, a characteristic cascade of biomarker changes emerged, starting with Aβ deposition more than two decades prior to onset, followed by metabolic dysfunction, hippocampal atrophy, and cognitive changes. Recently, enough longitudinal data has amassed on multiple biomarkers to start plotting individual trajectories with sufficient statistical power. In keeping with the cross-sectional data, an imaging study reported earlier this year pinpointed the first signs of Aβ deposition and metabolic dysfunction to the precuneus, followed by hippocampal atrophy (Feb 2018 news).
Now, Bateman and colleagues add longitudinal measurements of CSF Aβ42, total tau, and phospho-tau, as well as cognitive data. First author Eric McDade presented the bulk of the findings at the Alzheimer’s Association International Conference in London last year (Aug 2017 conference news).
The study analyzed data from 411 participants, including 251 mutation carriers. At each multiday visit, volunteers had PET and MRI scans to measure cortical Aβ, glucose metabolism, and atrophy; underwent lumbar punctures to assess CSF Aβ42, p-tau, and tau; and took a series of cognitive tests. Asymptomatic volunteers visited every two to three years, then every year after symptoms emerged. At baseline, volunteers were an average of 7.9 years away from estimated onset of symptoms, and just under half of all participants already had clinical symptoms of AD. Two hundred and seventeen people had at least one follow-up visit, with an average follow-up time of 2.7 years. Though the number of measurements taken varied across biomarkers, less than 10 people had more than five measurements for most markers.
The researchers plotted serial biomarker values against the volunteers’ estimated years from onset. Using these serial measurements, they could then estimate the annual rate of change in each biomarker across the course of disease—from 25 years prior to 10 years after estimated onset, comparing mutation carriers to noncarriers.
McDade and colleagues found that rates of Aβ deposition—according to cortical PiB uptake or CSF Aβ42—accelerated in carriers around 25 years before symptom onset, in keeping with widely reported data from studies of sporadic AD and other familial cohorts. Declines in precuneus glucose began around 17 years prior to onset, accelerating rapidly afterwards. Performance on some cognitive tests waned from three years prior to onset, while the hippocampus started to shrivel two years before, with shrinkage accelerating dramatically after onset.
While changes in most biomarkers moved in the same direction throughout the course of disease, there were two exceptions: total and phospho-tau in the CSF. The longitudinal data showed that both markers started off high, indicating levels were already elevated in most carriers prior to their baseline visit. Then, total tau held steady even after disease onset, while p-tau plummeted dramatically about five years before onset. These trajectories are at odds with earlier cross-sectional data from sporadic and familial AD, which indicate that total and phospho-tau in the CSF climb steadily in the years before onset. In fact, when McDade and colleagues analyzed the data from their cohort in a cross-sectional fashion, they, too, found t-tau and p-tau levels started climbing in mutation carriers relative to noncarriers at 14 and 11 years prior to estimated onset, respectively.
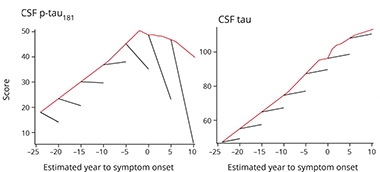
P-tau Plummet.
Rates of change in p-tau (left) and tau (right) rise steadily in the presymptomatic phase of AD according to cross-sectional analysis (red line), but remain level before plummeting (in the case of p-tau) when analyzed in five-year longitudinal stints. [Courtesy of McDade et al., Neurology, 2018.]
Why would cross-sectional and longitudinal data from the same cohort differ? A simple explanation is that CSF p-tau measurements varied widely between people, more so than any other marker. Cross-sectionally, this variability masked individual declines in CSF p-tau that occurred as symptoms approached. Looking at individual trajectories, it became clear that for mutation carriers with high p-tau, the marker tanked as symptoms approached, and plummeted even more steeply after onset. For noncarriers, levels of both tau and p-tau held steady throughout the timespan.
McDade hypothesized that the steep drop in CSF p-tau reflects its sequestration in neurofibrillary tangles. Preliminary tau PET data in the cohort support this idea, McDade said. But if falling CSF p-tau reflects the spread of tau tangles in the brain, what explains the jump in p-tau far earlier in the course of the disease? This is unclear, said McDade. Zetterberg proposed that early in the preclinical stages of disease, exposure to Aβ pathology triggers a dysregulation of tau, leading to truncation, phosphorylation, and active secretion of the protein. He pointed out that isotope-labeling studies suggest an active process of tau secretion in response to Aβ (Mar 2018 news). Later, tangles start to soak up the hyperphosphorylated tau, leading to a rapid decline in CSF p-tau concentrations that coincides with the onset of symptoms. This hypothesis will be put to the test as longitudinal tau PET data continue to mount in this cohort, both Zetterberg and McDade said.
While McDade emphasized that the longitudinal measurements most accurately reflect the dynamic biomarker changes that unfold during disease, Sebastian Palmqvist of Lund University in Sweden commented that this might not be true for all measures. “The longitudinal CSF p-tau finding is a bit counterintuitive and should be interpreted with caution,” he wrote. In particular, the conflicting trajectories of CSF p-tau estimated by the cross-sectional versus longitudinal data suggest that studies with more serial measurements are needed before solid conclusions are drawn, he said. The results also highlight the potential limitations of using longitudinal CSF p-tau as an outcome in clinical trials, given that the biomarker has an uncertain trajectory around the time of symptom onset.
McDade agreed, especially for trials testing therapies against tau. For example, a drug that targets neurofibrillary tangles might lead to a plateau, an increase, or simply slow the decline in CSF p-tau after symptom onset. While it is unclear whether CSF p-tau plummets in people with late-onset AD, McDade speculated that when neurofibrillary tangles spread out of the medial temporal lobe—a process that happens in both ADAD and LOAD—then such dynamics come into play. Zetterberg pointed out that due to the myriad biological processes potentially reflected by the CSF p-tau measurement, tau PET might make a more straightforward marker of tau pathology.
Finally, the findings support the idea that while Aβ accumulation sets the AD cascade in motion, it eventually becomes uncoupled from the disease process. This is demonstrated by the steady climb in Aβ deposition in the face of dramatic shifts in other biomarkers later in the disease, McDade pointed out. “If Aβ causes the disease, it happens relatively early on. Then at some point, the biomarkers take on an autonomous progression of their own,” he said. “This suggests that if we’re going to have any impact with Aβ-targeted therapies, then earlier is truly better.”
Pierre Tariot, Banner Alzheimer’s Institute in Phoenix, agreed. “These findings will undoubtedly impact the design of future preclinical trials in this population, in particular providing strong clues that, in future DIAN and Alzheimer’s Prevention Initiative (API) trials, we may need to intervene even earlier than we have thus far in the preclinical stage of illness.”—Jessica Shugart
References
News Citations
- DIAN Dispatch from Hawaii: Glimpse at Data, Push for Trials
- Paper Alert: DIAN Biomarker Data Show Changes Decades Before AD
- In Familial AD, Aβ Production Up, Clearance Down
- Aβ, Then Metabolism, Then Atrophy: In Familial AD, Cascade is Definitive
- Data from DIAN Revise Familiar Biomarker Trajectories
- Isotope Labeling Links Tau Production to Aβ Burden
Further Reading
Primary Papers
- McDade E, Wang G, Gordon BA, Hassenstab J, Benzinger TL, Buckles V, Fagan AM, Holtzman DM, Cairns NJ, Goate AM, Marcus DS, Morris JC, Paumier K, Xiong C, Allegri R, Berman SB, Klunk W, Noble J, Ringman J, Ghetti B, Farlow M, Sperling RA, Chhatwal J, Salloway S, Graff-Radford NR, Schofield PR, Masters C, Rossor MN, Fox NC, Levin J, Jucker M, Bateman RJ, Dominantly Inherited Alzheimer Network. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018 Oct 2;91(14):e1295-e1306. Epub 2018 Sep 14 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Banner Alzheimer's Institute
Yet another major contribution from DIAN. The investigators conducted a comprehensive longitudinal biomarker study in volunteers around the world who do or do not have autosomal dominant mutations that cause onset of Alzheimer’s symptoms at a generally early age, providing the best picture yet of the temporal sequence of pathological brain changes in autosomal dominant AD (ADAD). Annual rates of Aβ deposition measured by PET distinguished carriers from noncarriers nearly 25 years before the onset of AD symptoms. These were followed by changes in regional glucose metabolic rates, measured by PET, about 10 years later, and, even later, greater regional atrophy measured by MRI, followed by eventual cognitive decline. These findings generally map onto cross-sectional data reported previously by DIAN as well as by the Alzheimer’s Prevention Initiative (API) Colombia collaborative. However, the longitudinal data indicate that changes in measures of tau appear to occur closer to the time of clinical onset than suggested by cross-sectional data, an inconsistency that underscores the need for further studies.
The report overall provides the most detailed and compelling information yet about the progression of AD pathology in ADAD and supports the notion, also suggested by cross-sectional data, that measurable amyloid dysregulation precedes and possibly initiates neurodegeneration and cognitive decline in ADAD, not unlike the sequence reported in persons at elevated risk for late onset AD. These findings will undoubtedly impact the design of future preclinical trials in this population, in particular providing strong clues that, in future DIAN and API trials, we may need to intervene even earlier than we have thus far in the preclinical stage of illness. To this point, API reported at the 2018 AAIC that, in its preclinical trial in Colombia, a substantial proportion of ADAD mutation carriers lacked measurable Aβ deposition at baseline, a predefined goal in view of our collective uncertainty as to the predictive utility of this biomarker measure. That subgroup may ultimately prove to be especially informative.
Lund University
This study provides unique and important results in that it combines both cross-sectional and longitudinal biomarker data to map the pathogenesis of autosomal dominant AD. It also shows that the advantage of using longitudinal measures differs depending on biomarker type.
The longitudinal CSF p-tau finding is a bit counterintuitive and should be interpreted with caution. Here, the p-tau rates are overall negative (and become even more negative with time), but still the p-tau levels increase with time cross-sectionally. This estimated rate of change in CSF p-tau needs to be replicated to make sure that it is not caused by statistical or preanalytical issues. It also highlights the possible limitation of using longitudinal CSF p-tau as outcome in clinical trials. Future studies tracking CSF biomarker changes over longer time periods with at least three lumbar punctures are probably needed to obtain even more reliable longitudinal data.
When it comes to amyloid PET, FDG-PET, CSF Aβ42, and atrophy measures, the study nicely shows that longitudinal measures can increase the biomarker sensitivity to detect earlier pathological starting points that cannot be identified cross-sectionally because of large interindividual variability within the normal biomarker range.
Make a Comment
To make a comment you must login or register.