Derived Human Microglia Manage Without Functional TREM2
Quick Links
In the March 29 Stem Cell Reports, researchers led by Frederick Livesey at the University of Cambridge in England described a new protocol for generating microglia-like cells from human stem cells. Compared with a handful of other recently published microglia recipes, this one yields maximal numbers of cells using a relatively simple process. The microglia expressed genes on par with primary microglia cultured directly from the brain, buddied up with neurons in coculture, and responded to inflammatory stimuli. In microglia generated from people harboring pathogenic mutations in TREM2, the immune receptor’s trafficking was profoundly altered, yet the microglia gobbled up bacteria and pumped out cytokines just fine. However, some researchers wondered if these cells were tested in a physiologically meaningful paradigm.
- A new protocol robustly generates microglia from iPSCs.
- Transcriptionally and phenotypically, the cells mimicked primary microglia.
- Mutations in TREM2 derailed trafficking of the protein in these cells, but they responded normally to basic stimuli.
The pivotal role of microglia in neurodegenerative disease makes the immune cells a key subject of study, but efforts to understand them outside of their physiological environment have been fraught with challenges. For one, the highly responsive cells are shifty by nature—removing them from the brain evokes almost immediate changes in gene expression (Jun 2017 news). “You only have to look at them funny and they’ll get aggressive,” Livesey joked. The cells are also sparse in the mouse brain, and mouse microglia can only approximate the characteristics of their human counterparts. An emerging strategy to sidestep these issues is to make human microglia from scratch. In recent years, researchers have developed a handful of protocols that generate microglia from human stem cells, including the induced pluripotent variety (iPSCs) (Jul 2016 conference news). Reasoning that the environment of the brain is essential for microglial development, some protocols utilize complex coculture systems to coax precursors into microglia (Haenseler et al., 2017; Takata et al., 2017). Others rely entirely on cocktails of specific growth factors to steer the cells toward microgliahood (Muffat et al., 2016; Abud et al., 2017).
First author Philip Brownjohn and colleagues went with the cocktail approach, first generating primitive macrophage precursors (PMPs) from iPSCs. These cells approximate the macrophage precursors that arise from erythro-myeloid precursors in the yolk sac during fetal development and that give rise to brain-resident microglia. Other researchers previously developed protocols to establish them in culture (Karlsson et al., 2008; van Wilgenburg et al., 2013). Brownjohn found that iPSCs expanded by 20- to 50-fold into PMPs over 80 days. Then, by adding in other factors, including granulocyte colony-stimulating factor (GM-CSF) and IL-34, to replicate the environment of the developing brain, the researchers steered the PMPs into microglia-like cells within roughly a week. Ninety-five percent of the cells expressed typical microglial markers, including Iba-1 and CD45, while almost 100 percent of them expressed TREM2.
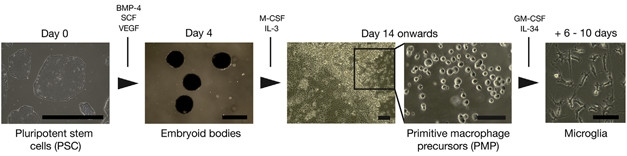
Making Microglia. Pluripotent stem cells grown in clumps (embryoid bodies) differentiated into primitive macrophage precursors and, finally, microglia. [Courtesy of Brownjohn et al., Stem Cell Reports, 2018.]
Brownjohn compared gene expression profiles to see how the derived cells measured up to primary microglia and to those developed via alternate methods (Zhang et al., 2016; Gosselin et al., 2017; Abud et al., 2017). Broadly, the transcriptome of the new microglia aligned well with those of other induced microglia, and of primary microglia that had been maintained in culture for a week. The transcriptome diverged from those reported for other types of myeloid cells such as monocytes and dendritic cells. While it also differed substantially from the transcriptome of freshly extracted microglia, it matched where it counts most: in expression of microglial signature genes (Hickman et al., 2013; Butovsky et al., 2014; Bennett et al., 2016). Livesey told Alzforum that while it would be preferable to generate microglia exactly like those in the brain, it may not be possible to escape what he called the “culture artifact.” He believes the PMP-derived microglia are a useful model to study processes in the brain.
In keeping with the status of such professional phagocytes, the induced microglia efficiently engulfed bacterial particles. And, when doused with lipopolysaccharide, the cells ramped up expression of inflammatory cytokines, including IL-1β, TNF-α, and IL-6. To see if the cells would interact appropriately with neurons, the researchers added them to three-dimensional cortical organoids. The microglia migrated deeply into the organoids and set up shop, extending probing processes to monitor their new environs.

Make Yourself at Home.
Microglia in culture (left) migrate deep into three-dimensional cortical organoids (right), spread out evenly, and take on a ramified shape. [Courtesy of Brownjohn et al., Stem Cell Reports, 2018.]
In a first stab at using these microglia to investigate processes related to neurodegeneration, the researchers next turned their sights on TREM2. They generated microglia from four iPSC sources: a patient with frontotemporal dementia who carried two copies of the T66M mutation in TREM2; two unaffected family members who carried only one copy of the mutant gene; and one person who carried two copies of the W50C mutation that causes Nasu-Hakola disease (NHD). All of the iPSC lines appeared to differentiate into microglia-like cells as well as those carrying two normal copies of TREM2. Using antibodies specific for different regions of TREM2, the researchers found that neither the W50C nor T66M variant trafficked normally to the cell surface, where the extracellular N-terminal domain is often shed following cleavage via ADAM10 protease. This cleavage leaves behind a C-terminal stub. Lysates from microglia bearing homozygous T66M or W50C mutations were devoid of this C terminal fragment. Overall, the findings indicated that the mutations disrupted TREM2 trafficking and processing, in agreement with studies reporting reduced maturation of the mutant protein in a mouse microglial cell line and in human embryonic kidney cells (Kleinberger et al., 2014; Jul 2014 webinar).
How would this stunted maturation affect microglial responses? To address this, the researchers first provoked the T66M and W50C TREM2 microglia with lipopolysaccharide (LPS). They found no deficits in the inflammatory responses as compared with induced microglia carrying wild-type TREM2. Normal and mutant microglia also engulfed fluorescently labeled E. coli with equal gusto. A previous study reported that TREM2 facilitates the uptake of acetylated lipoproteins, along with associated Aβ, and that pathogenic TREM2 mutations or TREM2 deficiency impaired this process (Yeh et al., 2016). However, the mutant cells had no problem engulfing these lipoproteins. The results are seemingly at odds with previous studies reporting that the mutations affect phagocytic and other microglial functions (May 2017 news).
Brownjohn and Livesey told Alzforum that the findings point to compensatory mechanisms—myriad other phagocytic receptors, for example—that could make up for TREM2’s loss of function in the mutant microglia.
In a joint comment to Alzforum, Christian Haass of the German Center for Neurodegenerative Diseases and Dominik Paquet of University Hospital, both in Munich, agreed that compensatory mechanisms were likely afoot, pointing out that they, too, had observed similar compensatory effects when assessing antibody-mediated phagocytosis of Aβ plaques in TREM2-deficient cells (Jul 2016 news).
Damian Crowther at AstraZeneca in Cambridge, England, offered a cautionary note. He referred to LPS as the “nuclear option,” pointing out that it maxes out pro-inflammatory responses and even downregulates TREM2 expression. This could mask differences between TREM2 mutants and wild-type cells, he said. “Of greater interest in neurodegenerative disease might be the interaction of the TREM2 pathway with weaker TLR4 agonists, such as amyloid-containing debris (reviewed in Molteni et al., 2016),” he wrote.
Likewise, Oleg Butovsky of Brigham and Women’s Hospital in Boston said he would like to see how the transcriptomes of the microglial cells change in response to disease-relevant stimuli, such as material from dying neurons. He previously reported that resting microglia express a homeostatic gene-expression signature, which is rapidly lost upon stimulation of TREM2 by neuronal detritus (Feb 2015 news; Sep 2017 news). Whether the TREM2 mutant microglia developed by Brownjohn and colleagues are deficient in changing gears will be a crucial next question to address with this beautiful cellular model, he said.
Brownjohn and Livesey told Alzforum that these experiments are already in the works, assessing how the cells migrate and respond to multiple threats in coculture with neurons.
How do Brownjohn’s microglia measure up to cells developed by other protocols? The jury is still out on that, according to Haass and Paquet. “At the current—still pioneering— state of the field, independent studies confirming transferability of protocols, validity of cellular fates/transcriptomics, as well as applicability for disease research are crucial. The recent paper by the Livesey lab therefore is a welcome addition,” they wrote.—Jessica Shugart
References
News Citations
- What Makes a Microglia? Tales from the Transcriptome
- Induced Microglia Make Debut at Keystone Symposium
- Paper Alert: TREM2 Crucial for Microglial Activation
- TREM2 Helps Phagocytes Gobble Up Aβ Coated in Antibodies
- Microglia in Disease: Innocent Bystanders, or Agents of Destruction?
- ApoE and Trem2 Flip a Microglial Switch in Neurodegenerative Disease
Webinar Citations
Paper Citations
- Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, Chintawar S, Schnell C, Antel JP, Allen ND, Cader MZ, Wade-Martins R, James WS, Cowley SA. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Reports. 2017 Jun 6;8(6):1727-1742. PubMed.
- Takata K, Kozaki T, Lee CZ, Thion MS, Otsuka M, Lim S, Utami KH, Fidan K, Park DS, Malleret B, Chakarov S, See P, Low D, Low G, Garcia-Miralles M, Zeng R, Zhang J, Goh CC, Gul A, Hubert S, Lee B, Chen J, Low I, Shadan NB, Lum J, Wei TS, Mok E, Kawanishi S, Kitamura Y, Larbi A, Poidinger M, Renia L, Ng LG, Wolf Y, Jung S, Önder T, Newell E, Huber T, Ashihara E, Garel S, Pouladi MA, Ginhoux F. Induced-Pluripotent-Stem-Cell-Derived Primitive Macrophages Provide a Platform for Modeling Tissue-Resident Macrophage Differentiation and Function. Immunity. 2017 Jul 18;47(1):183-198.e6. PubMed.
- Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, Jaenisch R. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 2016 Nov;22(11):1358-1367. Epub 2016 Sep 26 PubMed.
- Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, Caraway CA, Fote GM, Madany AM, Agrawal A, Kayed R, Gylys KH, Cahalan MD, Cummings BJ, Antel JP, Mortazavi A, Carson MJ, Poon WW, Blurton-Jones M. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron. 2017 Apr 19;94(2):278-293.e9. PubMed.
- Karlsson KR, Cowley S, Martinez FO, Shaw M, Minger SL, James W. Homogeneous monocytes and macrophages from human embryonic stem cells following coculture-free differentiation in M-CSF and IL-3. Exp Hematol. 2008 Sep;36(9):1167-75. Epub 2008 Jun 11 PubMed.
- van Wilgenburg B, Browne C, Vowles J, Cowley SA. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One. 2013;8(8):e71098. Epub 2013 Aug 12 PubMed.
- Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, Vogel H, Steinberg GK, Edwards MS, Li G, Duncan JA 3rd, Cheshier SH, Shuer LM, Chang EF, Grant GA, Gephart MG, Barres BA. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 2016 Jan 6;89(1):37-53. Epub 2015 Dec 10 PubMed.
- Gosselin D, Skola D, Coufal NG, Holtman IR, Schlachetzki JC, Sajti E, Jaeger BN, O'Connor C, Fitzpatrick C, Pasillas MP, Pena M, Adair A, Gonda DD, Levy ML, Ransohoff RM, Gage FH, Glass CK. An environment-dependent transcriptional network specifies human microglia identity. Science. 2017 Jun 23;356(6344) Epub 2017 May 25 PubMed.
- Hickman SE, Kingery ND, Ohsumi TK, Borowsky ML, Wang LC, Means TK, El Khoury J. The microglial sensome revealed by direct RNA sequencing. Nat Neurosci. 2013 Dec;16(12):1896-905. Epub 2013 Oct 27 PubMed.
- Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014 Jan;17(1):131-43. Epub 2013 Dec 8 PubMed.
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, Weissman IL, Chang EF, Li G, Grant GA, Hayden Gephart MG, Barres BA. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016 Mar 22;113(12):E1738-46. Epub 2016 Feb 16 PubMed.
- Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
- Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron. 2016 Jul 20;91(2):328-40. PubMed.
- Molteni M, Gemma S, Rossetti C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm. 2016;2016:6978936. Epub 2016 May 18 PubMed.
Other Citations
Further Reading
Papers
- Low D, Ginhoux F. Recent advances in the understanding of microglial development and homeostasis. Cell Immunol. 2018 Jan 10; PubMed.
Primary Papers
- Brownjohn PW, Smith J, Solanki R, Lohmann E, Houlden H, Hardy J, Dietmann S, Livesey FJ. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem Cell Reports, March 29, 2018.
Annotate
To make an annotation you must Login or Register.
Comments
Biomedizinisches Centrum (BMC), Biochemie & Deutsches Zentrum für Neurodegenerative Erkrankungen (DZNE)
Institute for Stroke and Dementia Research (ISD), University Hospital, LMU Munich
Since it is notoriously difficult to investigate microglial function in human brains, and so-called microglial cell lines do not faithfully reproduce the characteristics of microglia isolated from brain (Butovsky et al., 2014), human microglia are an extremely important tool for understanding function and dysfunction of these cells in neurodegenerative disorders. Over the last 18 months, several protocols have been published describing the differentiation of human microglia-like cells from stem cells, such as iPSCs. The first report by the Jaenisch lab (Muffat et al., 2016) was followed by several similar approaches using altered protocols (Abud et al., 2017; Douvaras et al., 2017; Haenseler et al., 2017; Pandya et al., 2017). The major differences in these protocols are: the use of defined differentiation factors vs. coculture with human iPSC-derived brain cells, such as astrocytes; the use of embryoid bodies (EBs) vs. cell monolayers during the first differentiation phase yielding hematopoietic progenitor-like cells; and the base media and growth factors used for directed differentiation and specific time points. While this variability should help bring out the best possible protocol, it makes it quite hard for labs that are new to the field to decide which protocol to implement. In general, protocols using EBs and cocultures with other cell types are harder to transfer to a new lab, since small alterations in the handling of these crucial steps can have dramatic consequences on the fate and number of differentiated cells. At the current—still pioneering—state of the field, independent studies confirming transferability of protocols, validity of cellular fates/transcriptomics, as well as applicability for disease research are crucial.
The recent paper by the Livesey lab, therefore, is a welcome addition, because it validates and expands the work of Sally Cowley’s lab (Haenseler et al., 2017) describing a very similar protocol, which itself was derived from earlier work on macrophage differentiation from Cowley’s lab (van Wilgenburg et al., 2013). However, Brownjohn et al. go beyond generating and characterizing human microglia-like cells by applying them to investigate TREM2 biology and AD-associated mutations. Nevertheless, one always needs to keep in mind that such iPSC-derived human microglia, which are kept outside of their natural environment under artificial culture conditions, may have several limitations that could affect their phenotypes in vitro.
Livesey and colleagues demonstrate nicely that human microglia expressing the TREM2 variants T66M and W50C show reduced maturation of the full-length protein, which as a consequence results in reduced shedding and reduced formation of the TREM2 C-terminal fragment. This is fully consistent with previous findings in human CSF (Kleinberger et al., 2014), where no sTREM2 is found in Nasu-Hakola patients carrying the T66M mutation. Furthermore, very similar findings were also made upon overexpression of human TREM2 in cultured cells (Kleinberger et al., 2014; Kober et al., 2016; Park et al., 2015), and in a mouse model with the T66M variant knocked in via the CRISPR/Cas9 technology (Kleinberger et al., 2017). In contrast to these findings, Brownjohn et al. found no TREM2 genotype-specific differences in response to LPS. However, this compares short-term in vitro experiments using cultured microglia (Brownjohn et al.,) with long-term in vivo analyses in mice (Kleinberger et al., 2017). More surprising may be, however, the lack of a difference in phagocytosis and LDL uptake as we and others reported in mice and upon heterologous expression of TREM2 variants (Kleinberger et al., 2017; Yeh et al., 2016). As stated by the authors, this may be due to compensatory effects via changes in the expression of other phagocytic receptors. Indeed, we have shown that antibody-dependent plaque clearance is reduced upon loss of TREM2 (Xiang et al., 2016). However, increased antibody dose compensated for reduced plaque clearance. Compensation was obtained upon loss of TREM2 function via increased expression of Fcg-receptors and enhanced Syk signaling (Xiang et al., 2016). Therefore, loss-of-function phenotypes are likely masked by compensatory mechanisms, which, as the authors state, is compatible with the late onset of the disease. It would be interesting to see if genotype-dependent differences become more apparent when human microglia are investigated in an environment that drives the switch of microglia from a homeostatic state to a disease-associated state, since under these conditions genes involved in chemotaxis and phagocytosis are upregulated in wild-type TREM2 expressing cells, but blocked in T66M mutants (Butovsky et al., 2014; Krasemann et al., 2017; Mazaheri et al., 2017). Moreover, one needs to keep in mind that even in human microglia, the TREM2 variants failed to maturate and thus lost their biological function, although this may be difficult to quantitate under resting conditions.
References:
Abud EM, Ramirez RN, Martinez ES, Healy LM, Nguyen CH, Newman SA, Yeromin AV, Scarfone VM, Marsh SE, Fimbres C, Caraway CA, Fote GM, Madany AM, Agrawal A, Kayed R, Gylys KH, Cahalan MD, Cummings BJ, Antel JP, Mortazavi A, Carson MJ, Poon WW, Blurton-Jones M. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron. 2017 Apr 19;94(2):278-293.e9. PubMed.
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE, Fanek Z, Liu L, Chen Z, Rothstein JD, Ransohoff RM, Gygi SP, Antel JP, Weiner HL. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014 Jan;17(1):131-43. Epub 2013 Dec 8 PubMed.
Douvaras P, Sun B, Wang M, Kruglikov I, Lallos G, Zimmer M, Terrenoire C, Zhang B, Gandy S, Schadt E, Freytes DO, Noggle S, Fossati V. Directed Differentiation of Human Pluripotent Stem Cells to Microglia. Stem Cell Reports. 2017 Jun 6;8(6):1516-1524. Epub 2017 May 18 PubMed.
Haenseler W, Sansom SN, Buchrieser J, Newey SE, Moore CS, Nicholls FJ, Chintawar S, Schnell C, Antel JP, Allen ND, Cader MZ, Wade-Martins R, James WS, Cowley SA. A Highly Efficient Human Pluripotent Stem Cell Microglia Model Displays a Neuronal-Co-culture-Specific Expression Profile and Inflammatory Response. Stem Cell Reports. 2017 Jun 6;8(6):1727-1742. PubMed.
Kleinberger G, Brendel M, Mracsko E, Wefers B, Groeneweg L, Xiang X, Focke C, Deußing M, Suárez-Calvet M, Mazaheri F, Parhizkar S, Pettkus N, Wurst W, Feederle R, Bartenstein P, Mueggler T, Arzberger T, Knuesel I, Rominger A, Haass C. The FTD-like syndrome causing TREM2 T66M mutation impairs microglia function, brain perfusion, and glucose metabolism. EMBO J. 2017 Jul 3;36(13):1837-1853. Epub 2017 May 30 PubMed.
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Kober DL, Alexander-Brett JM, Karch CM, Cruchaga C, Colonna M, Holtzman MJ, Brett TJ. Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife. 2016 Dec 20;5 PubMed.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, Beckers L, O'Loughlin E, Xu Y, Fanek Z, Greco DJ, Smith ST, Tweet G, Humulock Z, Zrzavy T, Conde-Sanroman P, Gacias M, Weng Z, Chen H, Tjon E, Mazaheri F, Hartmann K, Madi A, Ulrich JD, Glatzel M, Worthmann A, Heeren J, Budnik B, Lemere C, Ikezu T, Heppner FL, Litvak V, Holtzman DM, Lassmann H, Weiner HL, Ochando J, Haass C, Butovsky O. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity. 2017 Sep 19;47(3):566-581.e9. PubMed.
Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, Krasemann S, Capell A, Trümbach D, Wurst W, Brunner B, Bultmann S, Tahirovic S, Kerschensteiner M, Misgeld T, Butovsky O, Haass C. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017 Jul;18(7):1186-1198. Epub 2017 May 8 PubMed.
Muffat J, Li Y, Yuan B, Mitalipova M, Omer A, Corcoran S, Bakiasi G, Tsai LH, Aubourg P, Ransohoff RM, Jaenisch R. Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med. 2016 Nov;22(11):1358-1367. Epub 2016 Sep 26 PubMed.
Pandya H, Shen MJ, Ichikawa DM, Sedlock AB, Choi Y, Johnson KR, Kim G, Brown MA, Elkahloun AG, Maric D, Sweeney CL, Gossa S, Malech HL, McGavern DB, Park JK. Differentiation of human and murine induced pluripotent stem cells to microglia-like cells. Nat Neurosci. 2017 May;20(5):753-759. Epub 2017 Mar 2 PubMed.
Park JS, Ji IJ, An HJ, Kang MJ, Kang SW, Kim DH, Yoon SY. Disease-Associated Mutations of TREM2 Alter the Processing of N-Linked Oligosaccharides in the Golgi Apparatus. Traffic. 2015 May;16(5):510-8. Epub 2015 Feb 24 PubMed.
van Wilgenburg B, Browne C, Vowles J, Cowley SA. Efficient, long term production of monocyte-derived macrophages from human pluripotent stem cells under partly-defined and fully-defined conditions. PLoS One. 2013;8(8):e71098. Epub 2013 Aug 12 PubMed.
Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, Feederle R, Knuesel I, Kleinberger G, Haass C. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016 Sep 1;8(9):992-1004. PubMed.
Yeh FL, Wang Y, Tom I, Gonzalez LC, Sheng M. TREM2 Binds to Apolipoproteins, Including APOE and CLU/APOJ, and Thereby Facilitates Uptake of Amyloid-Beta by Microglia. Neuron. 2016 Jul 20;91(2):328-40. PubMed.
AstraZeneca IMED Biotech Unit
This paper describes a highly efficient method for generating microglia-like primitive macrophage precursors from wild-type, and TREM2-mutant (NHD-linked), fibroblasts. The wild-type PMPs have surface markers that we expect for macrophages and microglia (e.g., Iba1, CD45, and TREM2) and transcriptomic analysis, particularly when considering microglia-enriched transcripts, groups them with other preparations of primary cultured microglia. Thus, reassuringly, the stem cell-derived microglia are responding to the absence of the brain environment in the same way as primary cultured cells; furthermore, the cells are shown to be capable of migrating deep into brain organoids, eventually adopting a ramified, surveillance phenotype.
The important impact of this study on translational neuroscience stems from the ability to make microglia from patients with neurological diseases and compare them with healthy controls. Here the team induced microglia from patients with hemizygous and homozygous destabilizing mutations in TREM2 and show a progressive failure of the protein to be delivered to the plasma membrane, essentially replicating the results of Kleinberger et al. (2014) and others. Accordingly, the normal regulated proteolytic degradation of surface-expressed wild-type TREM2, by ADAM10/17 followed rapidly by g-secretase, is disrupted for the abnormally trafficked variants. This probably indicates that an alternative pathway is available for microglia to clear immature TREM2 variants that are stalled on the secretory pathway.
Things get even more exciting when Brownjohn and colleagues start to probe how the stem cell-derived microglia behave in innate immune system paradigms. It is known that the TLR4 and TREM2 signalling pathways exhibit reciprocal inhibition, however, in the context of the “nuclear option” of LPS agonism the pro-inflammatory pathway is maximally stimulated, likely rapidly downregulating TREM2 expression even in the wild-type cells. Of greater interest in neurodegenerative disease might be the interaction of the TREM2 pathway with weaker TLR4 agonist, such as amyloid containing debris (reviewed in Molteni et al., 2016), that may actually stimulate both pathways. The finding that induced microglia with destabilising TREM2 variants phagocytose E. coli normally, and show only minimal deficits in acetylated-LDL phagocytosis, goes against some literature for cultured microglia; however, as noted by the authors, this reflects the complexity of the system, where many proteins can detect innate immune triggers, and a possibly overlapping set of proteins may also directly mediate subsequent phagocytosis.
These results go some way to informing a debate on whether TREM2 acts as the “eyes” of the microglia—allowing them to sense debris and pathogens and stimulate phagocytosis, but not necessarily taking part directly in the phagocytosis—or whether TREM2 may (also) be the “mouth” of the microglia directly mediating the uptake of material. NHD teaches us that signalling by the “eyes” is essential, because loss of the TREM2 co-receptor DAP12 is an alternative cause of the disease—imagine the microglia in NHD as being blindly active. So, failure to see differences in the phagocytosis assays between wild-type and TREM2-variant lines is less of a challenge to our understanding of NHD as the failure of the TREM2-variant cells to modulate innate immune interactions. The answer to a better understanding of the interactions of the TLR4 and TREM2 pathways may be in the choice of possible TLR4 ligands—those that are likely to be prevalent in neurodegenerative disease may be more informative.
References:
Kleinberger G, Yamanishi Y, Suárez-Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger-Weinzierl A, Mazaheri F, Tahirovic S, Lleó A, Alcolea D, Fortea J, Willem M, Lammich S, Molinuevo JL, Sánchez-Valle R, Antonell A, Ramirez A, Heneka MT, Sleegers K, van der Zee J, Martin JJ, Engelborghs S, Demirtas-Tatlidede A, Zetterberg H, Van Broeckhoven C, Gurvit H, Wyss-Coray T, Hardy J, Colonna M, Haass C. TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med. 2014 Jul 2;6(243):243ra86. PubMed.
Molteni M, Gemma S, Rossetti C. The Role of Toll-Like Receptor 4 in Infectious and Noninfectious Inflammation. Mediators Inflamm. 2016;2016:6978936. Epub 2016 May 18 PubMed.
Make a Comment
To make a comment you must login or register.