Cytokine Needed to Keep Parkinsonian Neurodegeneration at Bay?
Quick Links
Brain inflammation burgeons during many neurodegenerative diseases, but it remains unclear exactly how this process contributes to pathology. In the October 8 Cell, researchers led by Shohreh Issazadeh-Navikas at the University of Copenhagen, Denmark, report evidence that inflammatory dysfunction alone can precipitate neurodegeneration. The authors analyzed mice that lacked the anti-inflammatory cytokine interferon β (IFN-β). The animals developed widespread pathology that shared features with Parkinson’s disease and dementia with Lewy bodies, including the accumulation of α-synuclein. The authors traced this buildup to a breakdown in the cellular waste disposal process known as autophagy. The findings suggest a crucial role for IFN-β in maintaining neuronal homeostasis, Issazadeh-Navikas said.
IFN- β has not been tied to neuron maintenance previously. Even so, this anti-inflammatory protein has been used for decades to treat people with multiple sclerosis (MS), and it is known to affect transcription of numerous genes (see Der et al., 1998). In MS, IFN- β dampens T cell responses, keeping these immune cells from attacking the myelin that insulates neurons (see Yong et al., 1998). Previously, Issazadeh-Navikas and colleagues reported that knocking out IFN- β in mice with experimental autoimmune encephalomyelitis, a model of MS, worsened neuroinflammation (see Teige et al., 2003). This led the authors to wonder how IFN- β affects normal brain function, and whether it might play a role in neurodegenerative disease.
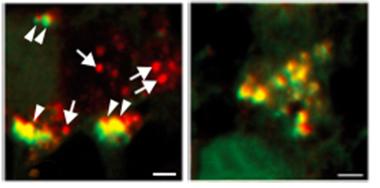
Autophagy Grinds to a Halt.
In cortical neurons from wild-type mice (left), autophagosomes (yellow) and autolysosomes (red) are both present, but the latter are rare in IFN-β knockout cells (right). [Courtesy of Cell, Ejlerskov et al.]
To investigate, first author Patrick Ejlerskov characterized IFN-β knockout mice (see Erlandsson et al., 1998). He found widespread neurodegeneration and discovered that it worsened with age. Starting at 1.5 months old, neurons withered at an accelerated rate in the hippocampus and olfactory bulb. By 6 and 12 months, respectively, death spread to the cerebellum and striatum. Knockouts generated fewer new neurons than wild-type, and their neurons had shorter, simpler processes. These brain changes were accompanied by behavioral deficits. From 3 months on, the knockouts struggled more than wild-types to balance on a spinning rod and cling to a wire. They reacted more strongly to pain, and had poorer spatial memory in water-maze tests.
To glean clues to what underlay these defects, the authors analyzed gene expression in knockout neurons using microarrays. They saw a boost in cell death pathways, as well as in genes linked to Parkinson’s, Huntington’s, and Alzheimer’s disease. In addition, many genes involved in autophagy were altered. The microarray data fit most closely with a Parkinson’s profile.
Experiments on cultured cortical and granular neurons from the IFN-β knockouts confirmed the microarray findings. In the normal autophagic process, autophagosomes containing waste proteins fuse with lysosomes, which digest the trash. Knockout neurons, however, possessed excess autophagosomes and very few autolysosomes, suggesting a block in the maturation of the latter (see image above). Knockout neurons also accumulated numerous aged, senescent mitochondria, implying a failure to dispose of these organelles as well.
Weak autophagy can cause many proteins to accumulate. The authors measured a build-up of phosphorylated α-synuclein, phospho-tau, and ubiquinated proteins. Because the gene-expression data hinted at a Parkinson’s phenotype, the authors examined the dopaminergic system in knockout mice. The animals lost dopaminergic neurons and accumulated α-synuclein deposits in Lewy bodies from 3 months on. Similar effects on autophagy and α-synuclein occurred in mice lacking the IFN-β receptor, IFNAR, strengthening the idea that the defects arose through this signaling pathway.
“The link between IFN-β and autophagy is one of the most interesting findings here,” said Mark Cookson at the National Institutes of Health, Bethesda, Maryland. He suggested that future studies investigate how IFN-β affects autophagy, and whether the mechanism is specific to IFN-β or occurs with knockout of other cytokines as well.
Neurons dispose of most of their α-synuclein through autophagy, so this protein is particularly prone to build up when autophagy slows down, Cookson added. Several genes associated with Parkinson’s, such as LRRK2, have been implicated in autophagy (see Jun 2012 news; Feb 2014 news).
Notably, degeneration in the IFN-β knockouts occurs without any mutation in known neurodegenerative proteins. “I think this opens a new window for looking at sporadic neurodegenerative diseases, and why they have associations with neuroinflammation,” Issazadeh-Navikas said. She wondered if perturbations in IFN-β signaling might contribute to the development of Parkinson’s, and plans to look for correlations between PD and genetic variants in IFN-β and related proteins.
If IFN-β speeds digestion of α-synuclein, could it help Parkinson’s patients? Some evidence from mice supports this idea. The authors injected lentivirus expressing the cytokine into the substantia nigra of rats that overexpress human α-synuclein (see Decressac et al., 2013). The treatment pumped up autophagy, preserved dopaminergic neurons, and improved motor control. However, delivery of the cytokine might pose problems for treating PD because it stays outside the blood-brain barrier. In MS treatment, IFN-β is given systemically and does not need to enter the brain. To promote autophagy, the cytokine needs to act directly on neurons.—Madolyn Bowman Rogers
References
News Citations
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- LRRK2 Interactions Identify New Parkinson’s Genes, Implicate Autophagy
Paper Citations
- Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998 Dec 22;95(26):15623-8. PubMed.
- Yong VW, Chabot S, Stuve O, Williams G. Interferon beta in the treatment of multiple sclerosis: mechanisms of action. Neurology. 1998 Sep;51(3):682-9. PubMed.
- Teige I, Treschow A, Teige A, Mattsson R, Navikas V, Leanderson T, Holmdahl R, Issazadeh-Navikas S. IFN-beta gene deletion leads to augmented and chronic demyelinating experimental autoimmune encephalomyelitis. J Immunol. 2003 May 1;170(9):4776-84. PubMed.
- Erlandsson L, Blumenthal R, Eloranta ML, Engel H, Alm G, Weiss S, Leanderson T. Interferon-beta is required for interferon-alpha production in mouse fibroblasts. Curr Biol. 1998 Feb 12;8(4):223-6. PubMed.
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Björklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A. 2013 May 7;110(19):E1817-26. Epub 2013 Apr 22 PubMed.
Further Reading
Primary Papers
- Ejlerskov P, Hultberg JG, Wang J, Carlsson R, Ambjørn M, Kuss M, Liu Y, Porcu G, Kolkova K, Friis Rundsten C, Ruscher K, Pakkenberg B, Goldmann T, Loreth D, Prinz M, Rubinsztein DC, Issazadeh-Navikas S. Lack of Neuronal IFN-β-IFNAR Causes Lewy Body- and Parkinson's Disease-like Dementia. Cell. 2015 Oct 8;163(2):324-39. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
U.K. Dementia Research Institute at University College London
The study by Ejlerskov et al. provides interesting new evidence for the role of Interferon-β (IFNβ) in the earliest events of the disease cascade of synucleinopathies such as Parkinson’s disease (PD) and dementia with Lewy bodies (DLB). In an attempt to elucidate the role of IFNβ in neurodegeneration, the authors analyze lfnb-/- mice, first testing for cognitive and behavioral deficits and then, upon observing a pronounced motor phenotype and behavioral deficits, determine a major role for the IFNβ/IFNAR pathway in neuronal autophagy.
The study has several important implications. First, the connection between proteins involved in the inflammatory system and neurodegeneration has been of great interest lately, but the common assumption has been that the mechanisms involved center around microglia-mediated clearance of amyloid protein. While the authors do not go into detail about whether this mechanism is contributing here (only glial counts, but not their activation state, were analyzed), they do provide another general cellular pathway, autophagy, that is affected by IFNβ/IFNAR and leads to dysregulation of protein homeostasis. Surprisingly, this dysregulation seems to lead to a motor phenotype and the neuronal aggregation of α-synuclein (αSyn). This is important, because secondly, the work further establishes a link between impaired autophagy, aggregation of αSyn, and early neurodegeneration in PD. Autophagy in general, and mitophagy in particular, have been strongly linked to PD through familial mutations in several genes encoding proteins such as PINK1/Parkin (Ashrafi et al., 2014) and LRRK2 (Schapansky et al., 2014; Orenstein et al., 2013). While αSyn has been shown to be degraded by chaperone-mediated autophagy in an in vitro system (Vogiatzi et al., 2008; Cuervo et al., 2004), the authors demonstrate here elegantly the accumulation of endogenous αSyn in an in vivo system when autophagy is impaired. The work is therefore interesting for a third reason, since it establishes a novel mouse model for PD that recapitulates the aggregation of αSyn in patient brain, without the need for overexpression of the protein itself, revealing pathways that are apparently upstream of amyloid and Lewy body formation. Now it will be important to study these pathways in this model in more detail to investigate whether they are linked not only to pathological changes in αSyn homeostasis but also to functional changes for other genes implicated in PD, as mentioned above.
In terms of changes in αSyn homeostasis, I find it very interesting that the authors report a decrease in physiological tetrameric αSyn, a form that has been found by us (Bartels et al., 2011) and others (Wang et al., 2011), while blockage of autophagy apparently leads to an accumulation of aggregation-prone monomeric αSyn. These changes recapitulate what is seen in SNCA mutation carriers (Dettmer et al., 2015) and therefore could link inherited PD cases to the underlying mechanism of sporadic PD, in which neuronal autophagy is potentially impaired by environmental factors. It is now important to investigate how the autophagy pathway could lead to an apparent shift in the ratio of tetrameric to monomeric αSyn, either through blockage of tetramer assembly pathways or the preferential degradation of one species over the other under these conditions.
Another open question is the specificity of αSyn accumulation versus other amyloidogenic proteins such as Aβ or prion protein that should also be affected by dysfunctional protein homeostasis. One could speculate that autophagosomal degradation is particularly important for αSyn degradation, but further studies are necessary. This study opens up a plethora of new ways to study the pathomechanism in PD and related neurodegenerative diseases and further raises the importance of the autophagic pathway as a target in drug discovery.
References:
Ashrafi G, Schlehe JS, LaVoie MJ, Schwarz TL. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J Cell Biol. 2014 Sep 1;206(5):655-70. Epub 2014 Aug 25 PubMed.
Schapansky J, Nardozzi JD, Felizia F, LaVoie MJ. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. 2014 Aug 15;23(16):4201-14. Epub 2014 Mar 27 PubMed.
Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013 Apr;16(4):394-406. Epub 2013 Mar 3 PubMed.
Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008 Aug 29;283(35):23542-56. PubMed.
Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science. 2004 Aug 27;305(5688):1292-5. PubMed.
Bartels T, Choi JG, Selkoe DJ. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature. 2011 Aug 14;477(7362):107-10. PubMed.
Wang W, Perovic I, Chittuluru J, Kaganovich A, Nguyen LT, Liao J, Auclair JR, Johnson D, Landeru A, Simorellis AK, Ju S, Cookson MR, Asturias FJ, Agar JN, Webb BN, Kang C, Ringe D, Petsko GA, Pochapsky TC, Hoang QQ. A soluble α-synuclein construct forms a dynamic tetramer. Proc Natl Acad Sci U S A. 2011 Oct 25;108(43):17797-802. Epub 2011 Oct 17 PubMed.
Dettmer U, Newman AJ, Soldner F, Luth ES, Kim NC, von Saucken VE, Sanderson JB, Jaenisch R, Bartels T, Selkoe D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun. 2015 Jun 16;6:7314. PubMed.
Make a Comment
To make a comment you must login or register.