Could Rogue APP Variants Invade Genome of Individual Neurons?
Quick Links
Multiple copies of the amyloid precursor gene crop up in adult neurons of the human brain, creating a heterogeneous neuronal landscape in a single person. This is especially true in brains from people with sporadic Alzheimer’s disease, but how this variation in amyloid precursor protein (APP), the precursor to the Aβ peptide, comes to be has remained a mystery. An article in the November 22 Nature proposes a mechanism and reveals a plethora of new forms of APP. Scientists led by Jerold Chun, Sanford Burnham Prebys Medical Discovery Institute, La Jolla, California, propose a DNA recombination process whereby mRNA from somatic cells is reverse-transcribed into complementary DNA (cDNA), which is then reinserted at some spot in the genome. Somewhere on the path between mRNA and cDNA reinsertion, APP exons disappear, point mutations pop up, and various insertions and deletions are introduced, creating thousands of APP variants in a single brain.
- APP mRNA is converted back to DNA and reinserted into the genome.
- Resulting “gencDNAs” are often mutated, common in sporadic AD.
- The process relies on the enzyme reverse transcriptase, inhibited in HIV/AIDS therapy.
This method of somatic gene recombination, if seemingly haphazard, allows neurons to mix up their genome content and diversify, the authors suggest. While the process takes place in healthy people and may even represent a mechanism for learning, it seems to intensify in sporadic AD. Intriguingly, 11 known APP mutations that cause familial AD were found in these mosaic neuronal genomes in people with sporadic disease.
“The study could alter our understanding of the roots of neurodegeneration,” wrote Guoliang Chai and Joseph Gleeson, both of the University of California, San Diego, in an accompanying News and Views. At the same time, they cautioned that it remains unknown whether this is a cause or an effect of disease, as did commentators contacted by Alzforum.
The paper drew a mixed response. “In this truly remarkable and ground-breaking study, the Chun group conclusively demonstrates a totally unexpected cause of genomic variability in the human brain,” wrote Christos Proukakis, University College London, who was not involved in the study.
Rachael Neve, Massachusetts General Hospital, Boston, cautioned that PCR and in situ hybridization are methods prone to artifacts. She would have liked to see data on more genes expressed outside the brain. “I would be more convinced of their hypothesis if some sort of control like that had been done,” she wrote to Alzforum. Chun said he plans to test more genes.

Cut and Paste.
After mRNA is transcribed from the APP gene and the introns spliced out, it can be reverse transcribed and inserted back into the genome, sometimes with glaring additions, omissions, or point mutations. [Courtesy of Chai and Gleeson, 2018. Nature.]
Neurons are among the few cell types in the body that have distinct genomes from one cell to the next, a phenomenon known as mosaicism (for a review, see Leija-Salazar et al., 2018; Verheijen et al., 2018). A few years ago, Chun and colleagues reported variable numbers of copies of APP in neurons from sporadic AD (SAD) patients (July 2015 news on Bushman et al., 2015).
In the present study, first author Ming-Hsiang Lee and colleagues took small, 50-neuron batches of neurons from the postmortem prefrontal cerebral cortices of three people who had had SAD and three elderly controls who had aged normally. The researchers amplified the APP transcripts and performed Sanger sequencing on them. As expected, they found the full-length APP-770 protein, as well as known splice variants APP-751 and APP-695. To their surprise, however, a host of other variants never before described turned up, as well. Twelve prominent ones stood out, but they were accompanied by many other, minor variants. Many were missing multiple central exons, meaning the two outer ends were pasted together. This created new intra-exonic junctions, or IEJs, new sequences that formed when two distant exons joined via complimentary sequences. The authors used these IEJs to identify these transcripts. Several of the RNA products could be translated and the products were toxic to SH-SY5Y cells.
Where were these RNA variants from? To find out if they came straight from the source—from DNA—Lee and colleagues sequenced APP fragments from the DNA of these cells. The DNA sequences exactly matched those found in the RNA. Unlike typical APP DNA, the variants lacked introns, suggesting that they had come from an RNA intermediate that had been reverse-transcribed into cDNA. The authors termed these regions genomic cDNA, gencDNA for short. These APP species seem specific to the brain, since some non-neuronal brain cells such as lung fibroblasts and human embryonic kidney cells showed little sign of them, though a comprehensive tissue comparison was not done. Neither did any gencDNAs for PSEN1 emerge, although that, too, was the only other gene the authors examined.
Lee and colleagues confirmed the presence of these gencDNAs in the genomes of six people with SAD and six without by DNA in situ hybridization—where probes specific for particular IEJs bound in the DNA. GencDNAs inserted at different spots in the genome, not necessarily near the original APP locus. A single neuron contained anywhere from zero to 13 gencDNAs.
To try to validate that these gencDNAs were real, the researchers used Agilent SureSelect DNA pulldown and short-read sequencing, an assay where APP variants are chopped up and the fragments sequenced, with overlapping nucleotides to help reassemble them into complete transcripts. All the gencDNAs analyzed lacked exon 8, which gets spliced from APP mRNA in the brain, further suggesting these species had their origins in mRNA.
To capture the diversity of APP gencDNAs, the researchers sequenced individual gencDNAs using single-molecule real-time (SMRT) circular consensus sequencing. In 96,424 neuronal nuclei isolated from five AD patients with sporadic AD, they found 6,299 unique APP sequences. By comparison, 162,245 nuclei from five elderly healthy controls yielded 1,084 different APP sequences. That gencDNAs are found in healthy brains suggests they may be part of some normal process, suggested Chun. However, neurons from SAD brains contain more gencDNA, with more SNVs, insertions, deletions, and novel IEJs, and much less of the normal APP transcript (see image below). Eleven gencDNAs from SAD patients were reported to contain autosomal-dominant AD mutations, including the Indiana, French, Austrian, Australian, and German mutations, which were absent in controls.
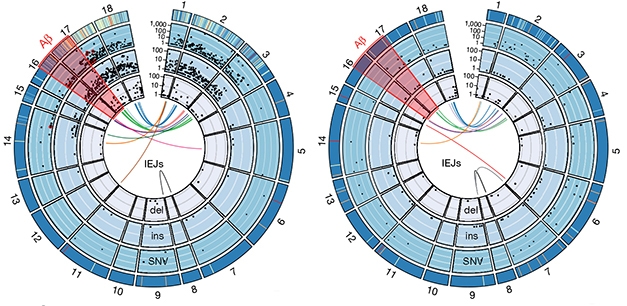
APP Diversifies. Exons 1–18 of APP (outer rim) contain thousands of single nucleotide variants (SNV), insertions (ins), and deletions (del), each represented by a black dot. Novel intra-exonic junctions (IEJs) connect disparate exons (colored lines in center). More gencDNAs invade neuronal DNA in SAD (left) than controls (right). Some contain known familial AD mutations (red dots). [Courtesy of Lee et al., 2018. Nature.]
Turning mRNA into cDNA requires the enzyme reverse transcriptase. Is this enzyme part of the process of creating gencDNA? The authors introduced an APP-containing plasmid into Chinese hamster ovary cells, which have endogenous RT activity. GencDNA appeared, but only if double-stranded breaks were created in the DNA first by hydrogen peroxide. When reverse transcriptase was inhibited, no gencDNA arose. That suggests to Chun that both reverse transcriptase and double-stranded DNA breaks are required for the generation of gencDNA.
In neuronal nuclei from J20 mice, which overexpress APP with the Swedish and Indiana mutations, the number of gencDNAs increased over a mouse lifetime of 2.3 years, suggesting accumulation with age.
This process of somatic APP gene recombination could enable a sort of recording and playback mechanism, wrote the authors, whereby the cell stores a preferred version of the APP transcript, sans introns, and can quickly access it later without having to splice it again. However, reverse transcriptase is a sloppy enzyme, hence the numerous alterations, Chun said. It’s possible that inhibiting this enzyme could prevent formation of gencDNAs, he added. Such inhibitors are FDA-approved for use in people with HIV.
“This study provides a potential mechanism leading to Alzheimer’s disease,” wrote Proukakis. “This work also gives fundamental insights into the brain genome, which is clearly much more plastic than previously thought.”
“I’m convinced what they detect is really there in the samples,” wrote Simon Mead, University College Hospital, London, to Alzforum. Mead praised the study as a fascinating technical advance that warrants independent replication in larger series of diseased human brains and different neurodegenerative diseases. That said, he cautioned that the current data don’t demonstrate a robust association with disease. “I would expect, if this is a real causal mechanism, that GWAS might have thrown up genes involved in DNA repair or gene recombination.”
Michael Rafii, University of Southern California, Los Angeles, echoed the question of causation. He said the work raises the possibility that genetic recombination may play a role in AD while opening up new avenues in fundamental neuroscience research. “The observation of neuronal DNA recombination via reverse transcription not only provides a mechanistic explanation for the mosaicism that has been previously observed in neurons, but also allows for diversification of each neuron’s unique protein repertoire over its long lifespan,” he wrote to Alzforum.—Gwyneth Dickey Zakaib
References
News Citations
Research Models Citations
Paper Citations
- Leija-Salazar M, Piette C, Proukakis C. Review: Somatic mutations in neurodegeneration. Neuropathol Appl Neurobiol. 2018 Apr;44(3):267-285. Epub 2018 Feb 28 PubMed.
- Verheijen BM, Vermulst M, van Leeuwen FW. Somatic mutations in neurons during aging and neurodegeneration. Acta Neuropathol. 2018 Jun;135(6):811-826. Epub 2018 Apr 28 PubMed. Correction.
- Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, Rehen SK, Yung YC, Chun J. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer's disease brains. Elife. 2015 Feb 4;4 PubMed.
Further Reading
Papers
- Nhan HS, Chiang K, Koo EH. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol. 2015 Jan;129(1):1-19. Epub 2014 Oct 7 PubMed.
- Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013 Jul 5;341(6141):1237758. PubMed.
- Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A. 2001 Nov 6;98(23):13361-6. PubMed.
Primary Papers
- Lee MH, Siddoway B, Kaeser GE, Segota I, Rivera R, Romanow WJ, Liu CS, Park C, Kennedy G, Long T, Chun J. Somatic APP gene recombination in Alzheimer's disease and normal neurons. Nature. 2018 Nov;563(7733):639-645. Epub 2018 Nov 21 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
UCL
In this truly remarkable ground-breaking study, the Jerold Chun group conclusively demonstrate a totally unexpected cause of genomic variability in the human brain: the incorporation of abnormal forms of the APP gene apparently reverse-transcribed from mRNA, missing introns, with novel inter-exonic junctions, and “point mutations” found in familial Alzheimer’s. The study uses numerous methods to support the findings, which are restricted to neurons and less prevalent or absent in controls, and convincingly shows an increase in these novel DNA insertions with age in a mouse model.
This study provides a potential mechanism leading to Alzheimer’s disease, which could even apply to other age-related neurodegenerative disorders, like PD, and studies in these will need to consider this possibility. This work also gives fundamental insights into the brain genome, which is clearly much more plastic than previously thought. The lead author has been a pioneer in this field for over 25 years, and this is building on the previous demonstration of increased APP exon copies in Alzheimer’s neurons, which have now been explained, mosaic aneuploidy, and common “conventional” CNVs arising in development of mouse neurons. Further work is needed to show whether other genes are affected, as presenilin-1 was not, or whether this phenomenon is mostly restricted to APP, and why this would be so. The authors even highlight the potential of anti-retroviral drugs to combat this attack on the neuronal genome.
University of Southern California Keck School of Mediicine
In a very thoughtful and well-designed set of experiments, Jerold Chun’s team further advances our knowledge of neuronal mosaicism and its possible relevance to brain function and disease. DNA replication must be extremely well-controlled and precise in order to facilitate accurate transfer of genetic information during cell division. Yet in the brain, where cell division occurs on a very limited basis, the observation of neuronal DNA recombination via reverse transcription not only provides a mechanistic explanation for the mosaicism that has been previously observed in neurons, but also allows for diversification of each neuron’s unique protein repertoire over its long lifespan.
Besides the discovery of a potential mechanism for neuronal DNA recombination, the finding that this mechanism is linked to neuronal activity and may play a role in AD pathogenesis is intriguing. In Down's syndrome (DS), chromosomal mosaicism resulting in aneuploidy of chromosome 21 (acquisition of a third copy of the APP gene), is thought to be the main pathogenic pathway to AD. Indeed, individuals with DS who are disomic for APP do not develop AD. Whether APP mosaicism is a cause for, or consequence of, AD pathogenesis in the sporadic population remains to be definitively established, but the work of Chun's lab raises the possibility that genetic recombination may play an important role in AD while opening up new avenues in fundamental neuroscience research.
University Maastricht
This exciting paper describes a new mechanism for generating genetic diversity in the brain, apparently acquired during life, especially in Alzheimer’s disease, and provides insight into how this may be related to neurodegenerative disease. It is a next view on the RNA world (e.g. Wang et al., 2014; Gout et al., 2017).
Accumulating evidence indicates that the brain is a genetic mosaic. Cell-to-cell genomic differences, which appear to be the result of somatic mutations during development and aging, contribute to cellular diversity in the nervous system. Interestingly, it has been shown that this mosaicism can also contribute to diseases of the brain (Verheijen et al., 2018). How these somatic mutations arise in nervous tissue remains largely unknown.
Previous work by the same group has found that neurons in sporadic AD patient brains contain more copies of APP (Bushman et al., 2015), a gene demonstrated to be causally involved in autosomal-dominant AD cases. Now, the authors provide a compelling explanation for this phenomenon: In their paper they show mosaic incorporation of many APP variants by error-prone reverse transcription, potentially resulting in toxic proteins and thereby contributing to sporadic AD. If confirmed by other research groups, this data adds a new layer of complexity to AD and potentially to neurobiology in general.
The study raises many questions:
The findings by Lee et al. provide a basis for new work on AD biology, a field that desperately needs new ideas (Morris et al., 2018).
References:
Wang IX, Core LJ, Kwak H, Brady L, Bruzel A, McDaniel L, Richards AL, Wu M, Grunseich C, Lis JT, Cheung VG. RNA-DNA differences are generated in human cells within seconds after RNA exits polymerase II. Cell Rep. 2014 Mar 13;6(5):906-15. Epub 2014 Feb 20 PubMed.
Gout JF, Li W, Fritsch C, Li A, Haroon S, Singh L, Hua D, Fazelinia H, Smith Z, Seeholzer S, Thomas K, Lynch M, Vermulst M. The landscape of transcription errors in eukaryotic cells. Sci Adv. 2017 Oct;3(10):e1701484. Epub 2017 Oct 20 PubMed.
Verheijen BM, Vermulst M, van Leeuwen FW. Somatic mutations in neurons during aging and neurodegeneration. Acta Neuropathol. 2018 Jun;135(6):811-826. Epub 2018 Apr 28 PubMed. Correction.
Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, Rehen SK, Yung YC, Chun J. Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer's disease brains. Elife. 2015 Feb 4;4 PubMed.
van Leeuwen FW, de Kleijn DP, van den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Köycü S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hol EM. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998 Jan 9;279(5348):242-7. PubMed.
De Strooper B, Karran E. The Cellular Phase of Alzheimer's Disease. Cell. 2016 Feb 11;164(4):603-15. PubMed.
Morris GP, Clark IA, Vissel B. Questions concerning the role of amyloid-β in the definition, aetiology and diagnosis of Alzheimer's disease. Acta Neuropathol. 2018 Nov;136(5):663-689. Epub 2018 Oct 22 PubMed.
This groundbreaking article suggests that APP somatic recombination represents a novel pathogenic mechanism potentially contributing to Alzheimer’s disease. This work has extremely interesting implications for brain functional genomics in general. However, replication studies are needed and many questions remain.
As Lee et al. note, somatic DNA recombination has not been previously described in the human brain. Nevertheless, their findings fit into an emerging framework indicating neurodegeneration is characterized by increased neural genomic instability. Neuronal hyperploidy (DNA content higher than in normal somatic cells) is raised in the early to mid-stages of AD before dropping later in disease, consistent with aberrant cell cycle re-entry preceding neuronal death (Arendt et al., 2010; Yang et al., 2001). Elevated DNA damage is observed in the C9ORF72-linked amyotrophic lateral sclerosis (ALS) spinal cord (Walker et al., 2017). Aging, one of the strongest risk factors for AD, increases the neuronal burden of somatic DNA variants present in some but not all of an individual’s cells (Hébert et al., 2013; Lodato et al., 2018). Indeed this reflects the age-dependent APP gencDNA increase Lee et al. detected in the J20 mouse model.
Lee et al. mention that APP gencDNAs are similar to but distinct from transposable elements (TEs). Interestingly, neurodegeneration-associated TE reactivation has been described in animal models of pathological proteins and in human brain tissue from the diseases in which they aggregate. Examples include tau, AD, and progressive supranuclear palsy (Guo et al., 2018; Sun et al., 2018); TDP-43, ALS, and frontotemporal dementia (Douville et al., 2011; Krug et al., 2017; Prudencio et al., 2017).
The presence of myriad neuronal APP gencDNA variants could have a range of complex effects on AD pathogenesis. An obvious implication is that increased APP gencDNA copies could elevate expression of their encoded proteins, ultimately raising amyloid-β levels. One intriguing possibility is whether these variants contribute to the stereological progression of pathology observed in AD (Montine et al., 2012). This could be investigated by quantifying APP gencDNA burden in different brain regions.
The article raises other important questions. Given the apparent importance of DNA lesions for APP somatic recombination, what mechanisms drive increased DNA strand breaks observed in AD (Mullaart et al., 1990) or APP transgenic mice (Suberbielle et al., 2013), e.g., neuronal activity (Madabhushi et al., 2015) or impaired DNA repair (Katyal and McKinnon, 2008)? Are gencDNA-encoded proteins expressed in vivo? Given the critical role glia play in mediating AD genetic risk (Huang et al., 2017; Sims et al., 2017), do neuronal-glial interactions influence neuronal APP somatic recombination?
The broader implications are exciting. In the human brain under normal physiological conditions, how many genes undergo somatic recombination, and how frequently? If only a subset of genes do, what sets them apart? This phenomenon could endow the brain with an additional physiological layer of transcriptomic control, in which previous gene expression experience dynamically sculpts neuronal transcriptomes to reinforce specific cellular states most effective for specific neural networks.
Why did neural somatic recombination evolve? One fascinating speculation is that somatic recombination could provide a selective mechanism operating at the neuronal cell level, whereby a constellation of gencDNA variants are generated across a cell population, and neurons whose genomes most benefit their neural network survive and remain integrated.
References:
Arendt T, Brückner MK, Mosch B, Lösche A. Selective cell death of hyperploid neurons in Alzheimer's disease. Am J Pathol. 2010 Jul;177(1):15-20. PubMed.
Yang Y, Geldmacher DS, Herrup K. DNA replication precedes neuronal cell death in Alzheimer's disease. J Neurosci. 2001 Apr 15;21(8):2661-8. PubMed.
Walker C, Herranz-Martin S, Karyka E, Liao C, Lewis K, Elsayed W, Lukashchuk V, Chiang SC, Ray S, Mulcahy PJ, Jurga M, Tsagakis I, Iannitti T, Chandran J, Coldicott I, De Vos KJ, Hassan MK, Higginbottom A, Shaw PJ, Hautbergue GM, Azzouz M, El-Khamisy SF. C9orf72 expansion disrupts ATM-mediated chromosomal break repair. Nat Neurosci. 2017 Sep;20(9):1225-1235. Epub 2017 Jul 17 PubMed.
Hebert LE, Weuve J, Scherr PA, Evans DA. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology. 2013 May 7;80(19):1778-83. PubMed.
Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, Yang P, Chittenden TW, Hatem NE, Ryu SC, Woodworth MB, Park PJ, Walsh CA. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018 Feb 2;359(6375):555-559. Epub 2017 Dec 7 PubMed.
Sun W, Samimi H, Gamez M, Zare H, Frost B. Pathogenic tau-induced piRNA depletion promotes neuronal death through transposable element dysregulation in neurodegenerative tauopathies. Nat Neurosci. 2018 Aug;21(8):1038-1048. Epub 2018 Jul 23 PubMed.
Douville R, Liu J, Rothstein J, Nath A. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol. 2011 Jan;69(1):141-51. PubMed.
Krug L, Chatterjee N, Borges-Monroy R, Hearn S, Liao WW, Morrill K, Prazak L, Rozhkov N, Theodorou D, Hammell M, Dubnau J. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017 Mar;13(3):e1006635. Epub 2017 Mar 16 PubMed.
Prudencio M, Gonzales PK, Cook CN, Gendron TF, Daughrity LM, Song Y, Ebbert MT, van Blitterswijk M, Zhang YJ, Jansen-West K, Baker MC, DeTure M, Rademakers R, Boylan KB, Dickson DW, Petrucelli L, Link CD. Repetitive element transcripts are elevated in the brain of C9orf72 ALS/FTLD patients. Hum Mol Genet. 2017 Sep 1;26(17):3421-3431. PubMed.
Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW, Duyckaerts C, Frosch MP, Masliah E, Mirra SS, Nelson PT, Schneider JA, Thal DR, Trojanowski JQ, Vinters HV, Hyman BT, . National Institute on Aging-Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol. 2012 Jan;123(1):1-11. PubMed.
Mullaart E, Boerrigter ME, Ravid R, Swaab DF, Vijg J. Increased levels of DNA breaks in cerebral cortex of Alzheimer's disease patients. Neurobiol Aging. 1990 May-Jun;11(3):169-73. PubMed.
Suberbielle E, Sanchez PE, Kravitz AV, Wang X, Ho K, Eilertson K, Devidze N, Kreitzer AC, Mucke L. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat Neurosci. 2013 May;16(5):613-21. PubMed.
Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, Stott RT, Gjoneska E, Nott A, Cho S, Kellis M, Tsai LH. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell. 2015 Jun 18;161(7):1592-605. Epub 2015 Jun 4 PubMed.
Huang KL, Marcora E, Pimenova AA, Di Narzo AF, Kapoor M, Jin SC, Harari O, Bertelsen S, Fairfax BP, Czajkowski J, Chouraki V, Grenier-Boley B, Bellenguez C, Deming Y, McKenzie A, Raj T, Renton AE, Budde J, Smith A, Fitzpatrick A, Bis JC, DeStefano A, Adams HH, Ikram MA, van der Lee S, Del-Aguila JL, Fernandez MV, Ibañez L, International Genomics of Alzheimer's Project, Alzheimer's Disease Neuroimaging Initiative, Sims R, Escott-Price V, Mayeux R, Haines JL, Farrer LA, Pericak-Vance MA, Lambert JC, van Duijn C, Launer L, Seshadri S, Williams J, Amouyel P, Schellenberg GD, Zhang B, Borecki I, Kauwe JS, Cruchaga C, Hao K, Goate AM. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer's disease. Nat Neurosci. 2017 Aug;20(8):1052-1061. Epub 2017 Jun 19 PubMed.
Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, Martin ER, Grenier-Boley B, Heilmann-Heimbach S, Chouraki V, Kuzma AB, Sleegers K, Vronskaya M, Ruiz A, Graham RR, Olaso R, Hoffmann P, Grove ML, Vardarajan BN, Hiltunen M, Nöthen MM, White CC, Hamilton-Nelson KL, Epelbaum J, Maier W, Choi SH, Beecham GW, Dulary C, Herms S, Smith AV, Funk CC, Derbois C, Forstner AJ, Ahmad S, Li H, Bacq D, Harold D, Satizabal CL, Valladares O, Squassina A, Thomas R, Brody JA, Qu L, Sánchez-Juan P, Morgan T, Wolters FJ, Zhao Y, Garcia FS, Denning N, Fornage M, Malamon J, Naranjo MC, Majounie E, Mosley TH, Dombroski B, Wallon D, Lupton MK, Dupuis J, Whitehead P, Fratiglioni L, Medway C, Jian X, Mukherjee S, Keller L, Brown K, Lin H, Cantwell LB, Panza F, McGuinness B, Moreno-Grau S, Burgess JD, Solfrizzi V, Proitsi P, Adams HH, Allen M, Seripa D, Pastor P, Cupples LA, Price ND, Hannequin D, Frank-García A, Levy D, Chakrabarty P, Caffarra P, Giegling I, Beiser AS, Giedraitis V, Hampel H, Garcia ME, Wang X, Lannfelt L, Mecocci P, Eiriksdottir G, Crane PK, Pasquier F, Boccardi V, Henández I, Barber RC, Scherer M, Tarraga L, Adams PM, Leber M, Chen Y, Albert MS, Riedel-Heller S, Emilsson V, Beekly D, Braae A, Schmidt R, Blacker D, Masullo C, Schmidt H, Doody RS, Spalletta G, Jr WT, Fairchild TJ, Bossù P, Lopez OL, Frosch MP, Sacchinelli E, Ghetti B, Yang Q, Huebinger RM, Jessen F, Li S, Kamboh MI, Morris J, Sotolongo-Grau O, Katz MJ, Corcoran C, Dunstan M, Braddel A, Thomas C, Meggy A, Marshall R, Gerrish A, Chapman J, Aguilar M, Taylor S, Hill M, Fairén MD, Hodges A, Vellas B, Soininen H, Kloszewska I, Daniilidou M, Uphill J, Patel Y, Hughes JT, Lord J, Turton J, Hartmann AM, Cecchetti R, Fenoglio C, Serpente M, Arcaro M, Caltagirone C, Orfei MD, Ciaramella A, Pichler S, Mayhaus M, Gu W, Lleó A, Fortea J, Blesa R, Barber IS, Brookes K, Cupidi C, Maletta RG, Carrell D, Sorbi S, Moebus S, Urbano M, Pilotto A, Kornhuber J, Bosco P, Todd S, Craig D, Johnston J, Gill M, Lawlor B, Lynch A, Fox NC, Hardy J, ARUK Consortium, Albin RL, Apostolova LG, Arnold SE, Asthana S, Atwood CS, Baldwin CT, Barnes LL, Barral S, Beach TG, Becker JT, Bigio EH, Bird TD, Boeve BF, Bowen JD, Boxer A, Burke JR, Burns JM, Buxbaum JD, Cairns NJ, Cao C, Carlson CS, Carlsson CM, Carney RM, Carrasquillo MM, Carroll SL, Diaz CC, Chui HC, Clark DG, Cribbs DH, Crocco EA, DeCarli C, Dick M, Duara R, Evans DA, Faber KM, Fallon KB, Fardo DW, Farlow MR, Ferris S, Foroud TM, Galasko DR, Gearing M, Geschwind DH, Gilbert JR, Graff-Radford NR, Green RC, Growdon JH, Hamilton RL, Harrell LE, Honig LS, Huentelman MJ, Hulette CM, Hyman BT, Jarvik GP, Abner E, Jin LW, Jun G, Karydas A, Kaye JA, Kim R, Kowall NW, Kramer JH, LaFerla FM, Lah JJ, Leverenz JB, Levey AI, Li G, Lieberman AP, Lunetta KL, Lyketsos CG, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, Miller CA, Miller JW, Morris JC, Murrell JR, Myers AJ, O'Bryant S, Olichney JM, Pankratz VS, Parisi JE, Paulson HL, Perry W, Peskind E, Pierce A, Poon WW, Potter H, Quinn JF, Raj A, Raskind M, Reisberg B, Reitz C, Ringman JM, Roberson ED, Rogaeva E, Rosen HJ, Rosenberg RN, Sager MA, Saykin AJ, Schneider JA, Schneider LS, Seeley WW, Smith AG, Sonnen JA, Spina S, Stern RA, Swerdlow RH, Tanzi RE, Thornton-Wells TA, Trojanowski JQ, Troncoso JC, Van Deerlin VM, Van Eldik LJ, Vinters HV, Vonsattel JP, Weintraub S, Welsh-Bohmer KA, Wilhelmsen KC, Williamson J, Wingo TS, Woltjer RL, Wright CB, Yu CE, Yu L, Garzia F, Golamaully F, Septier G, Engelborghs S, Vandenberghe R, De Deyn PP, Fernadez CM, Benito YA, Thonberg H, Forsell C, Lilius L, Kinhult-Stählbom A, Kilander L, Brundin R, Concari L, Helisalmi S, Koivisto AM, Haapasalo A, Dermecourt V, Fievet N, Hanon O, Dufouil C, Brice A, Ritchie K, Dubois B, Himali JJ, Keene CD, Tschanz J, Fitzpatrick AL, Kukull WA, Norton M, Aspelund T, Larson EB, Munger R, Rotter JI, Lipton RB, Bullido MJ, Hofman A, Montine TJ, Coto E, Boerwinkle E, Petersen RC, Alvarez V, Rivadeneira F, Reiman EM, Gallo M, O'Donnell CJ, Reisch JS, Bruni AC, Royall DR, Dichgans M, Sano M, Galimberti D, St George-Hyslop P, Scarpini E, Tsuang DW, Mancuso M, Bonuccelli U, Winslow AR, Daniele A, Wu CK, GERAD/PERADES, CHARGE, ADGC, EADI, Peters O, Nacmias B, Riemenschneider M, Heun R, Brayne C, Rubinsztein DC, Bras J, Guerreiro R, Al-Chalabi A, Shaw CE, Collinge J, Mann D, Tsolaki M, Clarimón J, Sussams R, Lovestone S, O'Donovan MC, Owen MJ, Behrens TW, Mead S, Goate AM, Uitterlinden AG, Holmes C, Cruchaga C, Ingelsson M, Bennett DA, Powell J, Golde TE, Graff C, De Jager PL, Morgan K, Ertekin-Taner N, Combarros O, Psaty BM, Passmore P, Younkin SG, Berr C, Gudnason V, Rujescu D, Dickson DW, Dartigues JF, DeStefano AL, Ortega-Cubero S, Hakonarson H, Campion D, Boada M, Kauwe JK, Farrer LA, Van Broeckhoven C, Ikram MA, Jones L, Haines JL, Tzourio C, Launer LJ, Escott-Price V, Mayeux R, Deleuze JF, Amin N, Holmans PA, Pericak-Vance MA, Amouyel P, van Duijn CM, Ramirez A, Wang LS, Lambert JC, Seshadri S, Williams J, Schellenberg GD. Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer's disease. Nat Genet. 2017 Sep;49(9):1373-1384. Epub 2017 Jul 17 PubMed.
Merck Research Labs-Boston
Lee et al. describe somatic mosaicism leading to increased copy number of the APP gene as a novel mechanism for amplified Aβ synthesis. This finding is consistent with the impact of triplication of the APP gene causing AD in Down’s syndrome and is supportive of Aβ lowering for AD prevention. Novel APP derived genomic cDNAs (gencDNAs) are also described as a source of APP-derived proteins that may contribute to neuronal toxicity. Some of these novel toxic proteins are predicted to be independent of β- and γ-secretase activity and so would not be impacted by inhibitors of these enzymes. This important new APP-related pathway for cell toxicity requires substantially more validation in human AD patients, tissues, and relevant cell lines to understand its relative contribution to the overall neuropathological cascade leading to AD. The potential elevation of somatic mosaicism with aging to be a common mechanism of neurodegeneration deserves intensive investigation for possible mechanisms of therapeutic interventions.
Make a Comment
To make a comment you must login or register.