Could Restoring Deep Sleep by Jolting the Brain Ward Off Alzheimer’s?
Quick Links
People with Alzheimer’s disease have trouble sleeping and awaken frequently, but exactly why this happens is unclear. In the November 3 Science Translational Medicine, researchers led by Jeannie Chin, Baylor College of Medicine, Houston, home in on a problem in the thalamic reticular nucleus (TRN), a brain area that regulates deep sleep. TRN neurons became less active at each worsening Braak stage in people who had AD, and also in a mouse model of amyloidosis. A bit like people, these mice slept in fits and starts, missing out on deep sleep. Jolting this region via an inducible receptor restored the mice's sleep quality, increased their slow-wave sleep, and slowed their plaque deposition. The authors suggest that if a way can be found to boost the TRN's activity, it might be therapeutic in AD.
- The thalamic reticular nucleus maintains sleep, regulates slow-wave activity.
- In people, neuron activity in this region dims with higher Braak stages.
- Could TRN activity be enhanced to treat Alzheimer's?
“This study is exciting because it directly shows that manipulating a specific sleep-related brain circuit can restore sleep and reduce Aβ plaques,” Laura Lewis of Boston University wrote to Alzforum. Luigi de Gennaro, University of Rome, Italy, agreed. “This work suggests a causal role of the TRN in mediating sleep and AD risk,” he wrote (full comment below).
Previously, Chin and colleagues noticed that J20 mice slept intermittently, and that their thalamic reticular nuclei were less active than those of control mice (Hazra et al., 2016). The TRN consists of a thin sheet of mostly inhibitory GABA-ergic neurons that surround the thalamus and block sensory input to enable undisturbed snoozing (Steriade and Timofeev, 2003; Fogerson and Huguenard, 2016; Lewis et al., 2015). “The TRN plays a critical role in coordinating network oscillations during sleep that are thought to be important for its beneficial effects on memory,” Lewis wrote.
Did activity dwindle in this region in people too? First author Rohan Jagirdar immunostained TRN tissue from 10 people who had had AD, three people who had had mild cognitive impairment, and 15 age-matched controls from the NIH NeuroBioBank. Compared to controls, neurons in diseased tissue expressed less FosB/∆FosB, a marker thought to reflect a history of neuronal activity. This marker tracked with Braak stage (see image below).

Worse Tauopathy, Quieter Neurons. TRN tissue from people with AD (red) or MCI (blue) expressed less of the neuronal activity marker FosB/ΔFosB than controls (gray). This correlated with Braak stage. [Courtesy of Jagirdar et al., Science Translational Medicine, 2021.]
Similarly, the researchers detected less FosB/ΔFosB in TRN tissue from J20 mice and two other sleep-disturbed models of amyloidosis, APP/PS1 and Tg2576, compared to controls. This suggested that impaired TRN activity may be a shared feature among AD and amyloidosis.
To see if the inactive TRN neurons affect sleep, the scientists video-monitored the mice's sleep and wake times in their cages. They implanted electrodes into the mice's frontal lobes and neck muscles to record electroencephalographs and electromyographs, respectively, as they slept. This was done with J20s only.
Compared to controls, 2-month-old J20 mice woke up twice as often, slept an hour less, and spent less time in slow-wave sleep. Synchronous firing of neurons during SWS helps convert short- to long-term memories and drain waste products from the brain (Jun 2015 news; Nov 2019 news). By 6 months, the mice awoke even more often, as amyloid plaques also developed.
Could making the TRN more active correct abnormal sleep? To address this question, the scientists used the “designer receptor exclusively activated by designer drug” (DREADD) method. In short, they injected a virus carrying a synthetic excitatory receptor into the TRN of 2-month-old mice, then implanted electrodes to measure EEG/EMG. To activate the receptor and jolt the TRN, the researchers fed the mice clozapine N-oxide (CNO; Roth, 2017). Mice manipulated in this way awoke less often, got more SWS, and had restored TRN neuron activity compared to animals given saline (see image below).
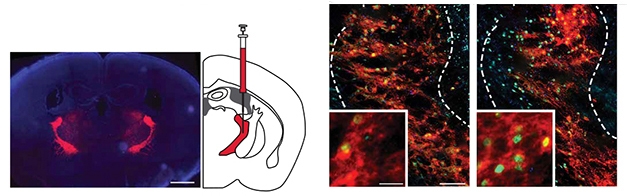
A DREADDed TRN. After injection of a virus encoding a CNO-inducible receptor labeled with mCherry dye (red), TRN tissue from mice given CNO (right) have more cells expressing FosB/ΔFosB (green) than mice given saline (left). [Courtesy of Jagirdar et al., Science Translational Medicine, 2021.]
To test if long-term TRN activation could reduce plaque load, Jagirdar injected 6-month-old mice with either CNO or saline before they fell asleep; they did this every day for a month and recorded EEG/EMG sleep data every five days. Treated mice slept more normally, expressed twice as much FosB/ΔFosB, and had 40 percent fewer plaques (see image below). “Daily, transient improvements in sleep are sufficient to produce measurable effects on AD-related pathology,” the authors conclude.

C NO Plaques. After a month of daily treatment, mice given CNO (right) had fewer plaques in the hippocampus and cortex than mice given saline (left). [Courtesy of Jagirdar et al., Science Translational Medicine, 2021.]
The scientists do not yet understand what exactly is silencing TRN neurons. They detected no plaques in the AD TRN tissue or tissue from 15-month-old J20 mice—or postmortem human TRN tissue. Chin’s lab is working to determine if the problem is intrinsic, such as low neuron excitability, or if an upstream input is impaired, causing the TRN to receive dimming signals. Chin noted that the deep cortical layers intimately interact with the TRN, so plaques there may clog communication.
These sleep studies were done in one model. Erik Musiek, Washington University, St. Louis, commented on the importance of studying sleep patterns and TRN activity in other amyloidosis models, as well. “These phenotypes can vary substantially between mouse lines, and the J20 mice have a neuronal hyperexcitability phenotype not seen in some other models,” he wrote to Alzforum. Lea Grinberg, University of California, San Francisco, pointed out that sleep processes differ between mice and humans, so this work may not be directly translatable. For example, Bryce Mander, University of California, Irvine, noted that the TRN primarily generates another type of brain wave called spindles during slumber. “I would have liked to see them explore what TRN disruption and stimulation does to sleep spindle expression,” he wrote (full comments below).
How could the TRN be stimulated in people? Chin and colleagues at the MD Anderson Neurodegeneration Consortium, Houston, are identifying differentially expressed genes in this region in J20 mice in search of potential targets, and are screening drugs against them. “Ideally, we envision a pill someone takes right before bed,” Chin said.
In the meantime, Chin does not think currently available sleep aid would help in the same way. Along with over-the-counter options, the orexin antagonist suvorexant is approved to treat insomnia in AD (Feb 2020 news; Oct 2014 news). Alas, Chin stressed the need to boost SWS specifically, as this is when the brain robustly clears debris. “I’m not sure the available products actually increase SWS,” she noted.
Chin plans to analyze postmortem TRN tissue from people with Parkinson’s disease or Down’s syndrome, which also come with sleep disturbances, to see if decreased TRN activity is common across multiple neurodegenerative conditions.
This paper is part of a special journal issue featuring articles on sleep, from how it influences brain waste clearance and blood flow to how neural patterns during slumber guide memory consolidation.—Chelsea Weidman Burke
References
Research Models Citations
News Citations
- Does Amyloid Disturb the Slow Waves of Slumber—and Memory?
- Deep Sleep Makes Waves for CSF
- Paper Alert: Phase 3 Trial Data Published on Sleep Aid Suvorexant
- Wake Up and Smell the … Orexin? Peptide Percolates in Alzheimer’s Brain
Therapeutics Citations
Paper Citations
- Hazra A, Corbett BF, You JC, Aschmies S, Zhao L, Li K, Lepore AC, Marsh ED, Chin J. Corticothalamic network dysfunction and behavioral deficits in a mouse model of Alzheimer's disease. Neurobiol Aging. 2016 Aug;44:96-107. Epub 2016 Apr 29 PubMed.
- Steriade M, Timofeev I. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron. 2003 Feb 20;37(4):563-76. PubMed.
- Fogerson PM, Huguenard JR. Tapping the Brakes: Cellular and Synaptic Mechanisms that Regulate Thalamic Oscillations. Neuron. 2016 Nov 23;92(4):687-704. PubMed.
- Lewis LD, Voigts J, Flores FJ, Schmitt LI, Wilson MA, Halassa MM, Brown EN. Thalamic reticular nucleus induces fast and local modulation of arousal state. Elife. 2015 Oct 13;4:e08760. PubMed.
- Roth BL. DREADDs for Neuroscientists. Neuron. 2016 Feb 17;89(4):683-94. PubMed.
External Citations
Further Reading
Papers
- Varga AW, Wohlleber ME, Giménez S, Romero S, Alonso JF, Ducca EL, Kam K, Lewis C, Tanzi EB, Tweardy S, Kishi A, Parekh A, Fischer E, Gumb T, Alcolea D, Fortea J, Lleó A, Blennow K, Zetterberg H, Mosconi L, Glodzik L, Pirraglia E, Burschtin OE, de Leon MJ, Rapoport DM, Lu SE, Ayappa I, Osorio RS. Reduced Slow-Wave Sleep Is Associated with High Cerebrospinal Fluid Aβ42 Levels in Cognitively Normal Elderly. Sleep. 2016 Nov 1;39(11):2041-2048. PubMed.
- Bubu OM, Brannick M, Mortimer J, Umasabor-Bubu O, Sebastião YV, Wen Y, Schwartz S, Borenstein AR, Wu Y, Morgan D, Anderson WM. Sleep, Cognitive impairment, and Alzheimer's disease: A Systematic Review and Meta-Analysis. Sleep. 2017 Jan 1;40(1) PubMed.
News
- Does Amyloid Accumulate After a Single Sleepless Night?
- Skimping on Sleep Makes For More Aβ in the Brain
- New Ties between AD and the Stages, Waves, and Molecules of Sleep
- Deep Sleep Makes Waves for CSF
- Does Amyloid Disturb the Slow Waves of Slumber—and Memory?
- Disturbed Sleep Exerts Toll on Memory and Neurodegeneration
- Do Brain Waves During Sleep Reflect Aβ and Tau Pathologies?
- Sleep Patterns, Circadian Clock Linked to Aβ Oxidative Stress
- From ApoE to Zzz’s—Does Sleep Quality Affect Dementia Risk?
- Rocking Improves Sleep and Memory in Adults
- Getting Too Little Sleep? You May Be Accumulating Amyloid.
- Tau, More than Aβ, Affects Sleep Early in Alzheimer’s
- Another Reason to Catch Some Zzzs: Sleep Regulates Tau Release
Primary Papers
- Jagirdar R, Fu CH, Park J, Corbett BF, Seibt FM, Beierlein M, Chin J. Restoring activity in the thalamic reticular nucleus improves sleep architecture and reduces Aβ accumulation in mice. Sci Transl Med. 2021 Nov 3;13(618):eabh4284. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Boston University
This compelling study shows that activating the TRN improves sleep and reduces Aβ in a mouse model of Alzheimer's disease. Disrupted sleep has long been thought to play a potential role in neurodegeneration, and this study is exciting because it directly shows that manipulating a specific sleep-related brain circuit can restore sleep and reduce Aβ plaques.
The TRN is a fascinating structure that is particularly effective at inducing the slow waves in neural activity that appear during non-rapid eye movement sleep, so it was a clever target for this study. Furthermore, bridging animal models with results in humans is often a challenge in the field.
An unusual aspect of this study is that they also look at the TRN from human patients with AD, and find that markers of TRN activity decline in AD and that this decline is correlated with Alzheimer's pathology. Future work could test whether this manipulation also affects cognitive abilities.
University of Rome
The last few years have seen mounting evidence on the role of sleep in relation to the risk of AD. Multiple lines of research indicate that sleep disruptions increase risk by altering production and/or clearance of disease-relevant proteins such as Aβ or tau. Aβ and tau concentrations in CSF fluctuate with the sleep-wake cycle, particularly with slow-wave sleep, in both humans and mice. On the other hand, SWS deprivation results in increased accumulation of both Aβ and tau. Some mechanisms by which disrupted sleep may increase the accumulation of such pathological proteins have been proposed or demonstrated: reduced clearance through the glymphatic system, alterations in lymphatic drainage, increased activity-dependent production of Aβ and tau. Furthermore, sleep fragmentation or poor sleep quality predict the risk of AD years before diagnosis. However, the precise mechanisms that give rise to sleep deficits in AD have yet to be fully clarified.
This very relevant study clearly points to a specific role of thalamic reticular nucleus, which has been previously demonstrated as essential for sleep maintenance and for the regulation of slow-wave sleep. The current study suggests a causal role of TRN in mediating the relation between sleep and risk of AD and playing a primary role in symptoms associated with AD, i.e., the burden of both Aβ and tau in the brain.
The study does not address in detail the possible role of cortical areas, though some studies have shown specific alterations there. For example, last April, we reported in a human study that people with AD and MCI compared to controls showed that (A) the cortical alterations of EEG during sleep (i.e., slowing of EEG frequencies) characterize both wake and REM sleep, (B) NREM sleep was associated with a reduction of 12-15 Hz EEG power in posterior cortical areas, (C) cortical EEG slowing in REM sleep was strongly correlated with cognitive impairment (D'Atri et al., 2021). All these findings point to an involvement of cortical areas, as well.
References:
D'Atri A, Scarpelli S, Gorgoni M, Truglia I, Lauri G, Cordone S, Ferrara M, Marra C, Rossini PM, De Gennaro L. EEG alterations during wake and sleep in mild cognitive impairment and Alzheimer's disease. iScience. 2021 Apr 23;24(4):102386. Epub 2021 Apr 1 PubMed.
Washington University School of Medicine
In this very interesting paper, Jagirdar et al. demonstrate both fragmented sleep and hyperactivity of the thalamic reticular nucleus in J20 APP transgenic mice. They use DREDDS to alter neuronal activity in this nucleus, thereby increasing slow-wave sleep and causing a modest but statistically significant decrease in amyloid plaques in the mice.
This finding implicates the TRN, a region known to regulate slow-wave sleep, in AD-related sleep dysfunction, providing novel neuroanatomical detail to our understanding of the relationship between sleep and amyloid deposition. This paper also strengthens the association between slow-wave sleep and amyloid accumulation.
It will be very important to evaluate both the sleep phenotype and TRN hyperactivity in other APP or APP/PS1 models of amyloid deposition, as these phenotypes can vary substantially between mouse lines. The J20 mouse in particular is known to have a neuronal hyperexcitability phenotype that is not seen in some other models, and which seems to precede plaque deposition. Since it is unclear if this is some effect of APP overexpression or of Aβ, confirmation in other APP models is key.
The authors have addressed this to some degree with FosB staining in the TRN of Tg2576 and APP/PS1 mice, which is an encouraging first step. Some interesting human postmortem FosB staining data is included in the paper, suggesting increased neuronal activity in the TRN of AD patients. This could open the door for electrophysiological or imaging studies to confirm this phenomenon in living AD patients, and to determine at what stage of the disease these changes occur.
University of California, San Francisco
This very elegant study uses a mixture of objective sleep assessment, chemogenetic manipulation, and electrophysiology of the thalamic reticular nuclei (TRN) in mice producing brain amyloidosis, one of the hallmarks of human AD. The authors did several experiments to establish that these mice show deficits of slow wave sleep (SWS) that correlate to levels of Aβ. Next, they established a role of TRN as a modulator of SWS, followed by acute and chronic manipulation of TRN, which resulted in improving SWS and lowering Aβ levels. Finally, the authors used postmortem human tissue and confirmed that TRN neurons have signs of being less active in AD than in controls in association with Braak stages.
This is an important study. It is a step forward in the effort to understand the mechanism underlying exacerbation of AD pathology in the context of chronic sleep dysfunction. Several studies show that sleep dysfunction increases the risk of accumulating Aβ and tau and the risk of dementia. This study also put the TRN in the spotlight as a possible key player modulating SWS in AD context. It is to be seen if the same connection is true in humans, as mice and humans have somehow different sleep mechanisms.
An intriguing question not answered by this study is what causes TRN deficits. This region is not prone to accumulate amyloid plaques in mice or human (tested in the study), nor tau (tested by Rüb et al., 2016).
Another intriguing follow-up question is why patients with PSP, a primary tauopathy that causes profound lack of sleep from early stages, don’t accumulate amyloid. It begs the question whether SWS dysfunction is the main reason behind exacerbated amyloid deposition in AD. Better animal models that also accumulate AD tau may shed light on this question.
References:
Rüb U, Stratmann K, Heinsen H, Turco DD, Seidel K, Dunnen Wd, Korf HW. The Brainstem Tau Cytoskeletal Pathology of Alzheimer's Disease: A Brief Historical Overview and Description of its Anatomical Distribution Pattern, Evolutional Features, Pathogenetic and Clinical Relevance. Curr Alzheimer Res. 2016;13(10):1178-97. PubMed.
University of California, Irvine
Overall, this nice series of experiments identifies yet another component of the sleep/wake regulatory system—the TRN—that is disrupted by Aβ and degenerated in AD.
It is interesting that stimulating the TRN can impact Aβ. It is unlikely, however, that the TRN is the only stimulation target necessary, and that stimulating it would address all the sleep disruptions caused by Aβ and also tau pathology.
AD pathophysiology impacts multiple different systems regulating sleep, including brainstem and hypothalamic nuclei, cortical nodes regulating slow wave expression, and even the hippocampus that regulates ripples, and coupling of slow waves, sleep spindles, and ripples.
This is an important experiment, and future studies need to address whether interventions to enhance TRN activity in humans can be helpful in reducing AD risk.
I would have liked to see the authors also explore what TRN disruption and stimulation does to sleep spindle expression, since sleep spindles, unlike slow waves, are the primary sleep feature generated in the TRN.
Washington University School of Medicine
This interesting study convincingly contributes to the growing literature on the importance of sleep in AD, particularly in the context of Aβ plaque pathology. The authors confirm several fundamental effects on reduced sleep and non-REM slow-wave sleep reduction associated with progressive accumulation of Aβ deposition (Kang et al., 2009; Roh et al., 2014; Mander et al., 2015; Lucey et al., 2018) due to increased neuronal activity (Holth et al., 2019). Furthermore, this study provides mechanistic insights into how GABAergic neuronal activity in the thalamic reticular nucleus (TRN) regulates sleep in an APP transgenic mouse model, which is further supported by observed reduction in neuronal activity marker, FosB, in postmortem tissue analysis in mild cognitive impairment and AD cases.
While the Chin lab had previously shown that TRN neurons exhibit reduced activity but without overt neuronal loss (Hazra et al., 2016), the authors investigated whether this may be a causal mechanism driving sleep fragmentation and reduced slow-wave sleep in APP mice with substantial plaque burden. The authors tested this hypothesis by selectively activating TRN neurons using DREADD in both non transgenic and APP hM3Dq mice along with saline or clozapine administration to activate GABAergic neuronal activity. Perhaps the most fascinating finding of the study is how treating APP hM3Dq mice with clozapine essentially rescued the reduction in slow-wave sleep activity and overall sleep fragmentation, at a comparable level to non-Tg hM3Dq mice treated with saline. In line with this finding, chronic activation of TRN neurons reduced plaque burden in APP mice, suggesting that improving slow-wave sleep activity may prove beneficial as a potential treatment for plaque pathology in AD.
Given the beneficial effect of slow-wave sleep on memory, it would be interesting to see whether increased TRN neuronal activity could potentially rescue behavioral impairments associated with impaired memory consolidation in APP mice.
References:
Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009 Nov 13;326(5955):1005-7. PubMed.
Roh JH, Jiang H, Finn MB, Stewart FR, Mahan TE, Cirrito JR, Heda A, Snider BJ, Li M, Yanagisawa M, de Lecea L, Holtzman DM. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer's disease. J Exp Med. 2014 Dec 15;211(13):2487-96. Epub 2014 Nov 24 PubMed.
Mander BA, Marks SM, Vogel JW, Rao V, Lu B, Saletin JM, Ancoli-Israel S, Jagust WJ, Walker MP. β-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015 Jul;18(7):1051-7. Epub 2015 Jun 1 PubMed.
Hazra A, Corbett BF, You JC, Aschmies S, Zhao L, Li K, Lepore AC, Marsh ED, Chin J. Corticothalamic network dysfunction and behavioral deficits in a mouse model of Alzheimer's disease. Neurobiol Aging. 2016 Aug;44:96-107. Epub 2016 Apr 29 PubMed.
Lucey BP, Hicks TJ, McLeland JS, Toedebusch CD, Boyd J, Elbert DL, Patterson BW, Baty J, Morris JC, Ovod V, Mawuenyega KG, Bateman RJ. Effect of sleep on overnight cerebrospinal fluid amyloid β kinetics. Ann Neurol. 2018 Jan;83(1):197-204. PubMed.
Holth JK, Fritschi SK, Wang C, Pedersen NP, Cirrito JR, Mahan TE, Finn MB, Manis M, Geerling JC, Fuller PM, Lucey BP, Holtzman DM. The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science. 2019 Feb 22;363(6429):880-884. Epub 2019 Jan 24 PubMed.
Dementia Research Institute at Imperial College London
The results of this paper seem robust and validated with good controls. Their APP model develops rapid sleep disturbances, mainly fragmentation, decreased slow-wave sleep, as well as total sleep time during the light (resting) phase. At the same time, these mice develop cognitive impairments. By activating the TRN with CNO injection (which selectively activates GABAergic neurons in the TRN, TRN being mainly populated by GABAergic neurons), they reverse the sleep disturbances. By chronically improving sleep via the TRN, they managed to alleviate Aβ pathology in the brain. Also, postmortem analysis of brain tissue of patients who suffered from Alzheimer and MCI indicated a decrease of activity in the TRN. There is no mechanism proposed, but I think it’s quite elegant.
The link between sleep and Aβ is well-documented. It has been shown that sleep loss is a risk factor for dementia. In addition, mounting evidence suggests that sleep disturbances are a prodromal phase of Alzheimer’s disease, and that pathological amyloid aggregates could be involved in sleep alterations. These two aspects are not mutually exclusive. However, the underlying mechanisms linking sleep, amyloid, and ultimately Alzheimer’s disease are poorly understood. Here, the authors show that the activity of the TRN, a brain region involved in sleep regulation, is impaired in a mouse model of Alzheimer’s disease exhibiting increased Aβ, as well as in the brains of patients suffering from the same condition. Furthermore, mice display sleep alterations. They show that by reversing the decreased neuronal activity of the TRN in mice, it improves sleep disturbances, as well as decreases amyloid plaques in the brain. This work represents an important steppingstone to better understand why sleep is disturbed in Alzheimer’s disease.
It would have been nice to see whether the TRN activation affected this mouse model’s known cognitive disruption. Also, we don’t know whether the patients suffering from Alzheimer's or MCI whose brain tissue was examined in this study also exhibited sleep disturbances.
Memory Clinic-NPZ
While this is an interesting study in animals, I would hesitate to extrapolate it to humans. A study we published as long ago as 1986 indicates that SWS in itself is an ambiguous electrophysiological phenomenon and not indicative of a specific brain state. Thus, just increasing EEG slow waves by any kind of manipulation may not lead to meaningful effects in human subjects.
References:
Spiegel R, Köberle S, Allen SR. Significance of slow wave sleep: considerations from a clinical viewpoint. Sleep. 1986;9(1):66-79. PubMed.
Make a Comment
To make a comment you must login or register.