Caught in the Act—Amyloid Damages Neurons
Quick Links
Since Alois Alzheimer’s first descriptions of senile plaques, we have known that they are associated with dementia. But are they detrimental? Are these amyloid-β (Aβ) deposits perpetrators or innocent bystanders? Nearly a century later, a verdict is still not in. Now that we know neurodegeneration can occur without plaque formation, and that intracellular Aβ (see ARF related news story) and small oligomers of the peptide may cause their fair share of toxicity (see ARF related news story), is it time to acquit the larger Aβ deposits? Maybe not—a report in the October 10 Nature Neuroscience online bolsters the case against plaques. The authors provide new photographic evidence implicating Aβ deposits in neuronal damage.
Wen-Biao Gan and colleagues at New York University School of Medicine used two-photon fluorescent imaging to examine neuronal architecture in the vicinity of plaques. First author Julie Tsai and coauthors found that in brain tissue from transgenic mice expressing mutant human amyloid-β precursor protein, neurons that passed through amyloid plaques were damaged. On average, dendrites in plaques had lost nearly 50 percent of their spines, the pinhead-like structures that facilitate communication between dendrites and other neurons. Axons that passed through, or skirted, plaques were also dramatically affected, becoming either constricted, or ballooning out like varicose veins. The danger zone appeared to extend to 15 micrometers from the plaque surface. Within this sphere, 90 percent of axons showed some degree of varicosity. Axons outside this boundary appeared normal.
Tsai and colleagues were able to extend these observations to living tissue. Using a recently developed transcranial microscope, they peered through the skulls of transgenic mice and were able to image amyloid deposits, and their surrounding neurons, for weeks at a time. Tsai and colleagues found that both axons and dendrites were gradually eliminated if they were within 15 micrometers of a plaque. In fact, over 36 percent of them were eliminated during a period of 4-5 weeks. The results indicate that neurodegeneration occurs after, or at least concomitantly with, plaque formation.
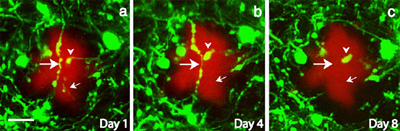
A picture paints a thousand words
These two-photon fluorescent images, taken transcranially from live mice reveal that neurites near amyloid deposits undergo gross morphological changes, and eventually disappear. In these images, taken over a period of eight days, neurons stained with the lipophilic dye DiI appear green, while amyloid plaques appear red (scale bar is 10 micrometers). [Image courtesy of Wen-Biao Gan and Nature Neuroscience]
This work supports similar findings that were published in the late nineties using fixed tissue sections from transgenic animals (see, for example, Phinney et al., 1999 and Knowles et al., 1999). “Our results suggest that the accumulation of fibrillar amyloid causes not only local structural disruption of synapses but also eventual neurite breakage, indicating that amyloid deposition is far more detrimental to the brain than previously thought,” write the authors, though they concede that the damaging factor may not be fibrillar Aβ alone, but could include small Aβ peptides.—Tom Fagan
References
News Citations
- Philadelphia: The Enemy Within—Neurodegeneration From Intraneuronal Aβ
- Oligomers in AD: Too Much of a Bad Thing?
Paper Citations
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M. Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci. 1999 Oct 1;19(19):8552-9. PubMed.
- Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT. Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci U S A. 1999 Apr 27;96(9):5274-9. PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Tsai J, Grutzendler J, Duff K, Gan WB. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat Neurosci. 2004 Nov;7(11):1181-3. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Pennsylvania
This is a very interesting paper that adds to our understanding of the in-vivo biology of plaques, and it would be interesting to add further nuances to these insights by comparing the effects of diffuse versus neuritic plaques on the integrity of dendrites, since in an earlier study >10 years ago of diffuse plaques in human cerebellum (Li et al., 1994), we did not see evidence of plaque toxicity on Purkinje cell dendrites, but in addition to the fact that we restricted our observations to diffuse plaques, the methods we used were diffrent from those reported here by Tsai et al. Nonetheless, given the many known differences between diffuse and neuritic plaques, it would be important to understand if these differences also extend to the relative toxic potential of each of these plaque types, since diffuse plaques may harbor components that mitigate toxicity which are not found in neuritic plaques.
References:
Li YT, Woodruff-Pak DS, Trojanowski JQ. Amyloid plaques in cerebellar cortex and the integrity of Purkinje cell dendrites. Neurobiol Aging. 1994 Jan-Feb;15(1):1-9. PubMed.
Columbia University
In studies quite similar to those of Tsai et al., we did not see clear associations between amyloid plaques and the loss of dendritic spines (Moolman et al., 2004), though we did a observe a similar (approximately 50 percent) loss of spines in two different mouse models of AD at 11 months of age and a suggestion of loss at eight months of age. We have tended toward the view that it is soluble Aβ rather than Aβ that is organized into plaques that are responsible for alterations in synaptic plasticity. It is certainly possible that soluble Aβ is higher in the region of the plaque, and that would reconcile the observations. Floyd Bloom's lab has pointed to dendritic alterations very early in the life of another AD model mouse prior to significant plaque formation, but did not specifically look at spines (Wu et al., 2004).
References:
Moolman DL, Vitolo OV, Vonsattel JP, Shelanski ML. Dendrite and dendritic spine alterations in Alzheimer models. J Neurocytol. 2004 May;33(3):377-87. PubMed.
Wu CC, Chawla F, Games D, Rydel RE, Freedman S, Schenk D, Young WG, Morrison JH, Bloom FE. Selective vulnerability of dentate granule cells prior to amyloid deposition in PDAPP mice: digital morphometric analyses. Proc Natl Acad Sci U S A. 2004 May 4;101(18):7141-6. PubMed.
Lund University
These interesting papers by Tsai et al. and Moolman et al. further support the idea that neuronal processes are important sites of damage during AD pathogenesis. A major question, of course, is why are processes being damaged? Is there any damage prior to plaques? Both papers consider the possibility that increases in levels of soluble Aβ may be important. In addition, increasing evidence supports a role for intraneuronal Aβ accumulation in the degeneration of processes (for example, see Wirths et al., 2004, for a current review).
University of California, San Francisco
I think the data in the Gan paper is quite beautiful. I especially
liked the time-lapse results in Figure 2 using the transcranial
approach. I think their findings nicely extend previous work that
showed an association between abnormalities of dendrites and the
proximity of plaques. I also am especially interested in the effects
on the rate of spine formation and elimination, since I think a lot of
the symptoms in AD probably arise from synaptic dysfunction rather
than neuronal death.
The work also raises a lot of intriguing questions. Is the effect
they see really due to the amyloid in the plaque or is the plaque a
major source for more toxic diffusible forms of Aβ whose
concentration is therefore high around the plaque? Either model would
explain the close relationship between the existence of dendritic
abnormalities and their distance from the plaque. However, the answer
would have big implications for developing therapeutic strategies. To
answer the question, they would need some way to simultaneously
visualize both forms of Aβ (plaque and diffusible) in living
tissue. Another question is whether the plaque-associated
abnormalities are a major or minor contributor to the overall
behavioral phenotype—that one is a lot tougher.
Hertie Institute for Clinical Brain Research
I agree with the previous comments in that this paper provides elegant confirmation of earlier research on how plaques impact neural structure. In addition, they show a substantial increase in the turnover of spines and dystrophic axon terminals, indicating that abnormal plasticity is occurring. Whether this is caused by amyloid or other diffusible molecules such as growth factors, this abnormality is likely to strain the capacity to produce, synchronize, and transport plasticity-related proteins—in addition to the obvious negative effects of the structural changes. It will, thus, also be interesting to see how this relates to the function of affected neural systems.
University of Concepcion
This paper shows a nice view of the neurodegeneration of dendritic spines, but I am not really sure how it relates plaques to mophological changes. In this experiment we would need examine the role of the glia cells, and their contribution to plaque formation. I wonder if it is possible to see the same effect without glial cells?
Make a Comment
To make a comment you must login or register.