C9ORF72, Sporadic ALS Transcriptomes Now Available
Quick Links
What goes wrong in the brain of a person with a hexanucleotide expansion in C9ORF72, a protein involved in RNA processing? Quite a bit, according to an analysis published in the July 20 Nature Neuroscience. Researchers from the Mayo Clinic profiled the brain transcriptomes of both C9ORF72-linked and sporadic cases of amyotrophic lateral sclerosis, and discovered that the expansion-toting brains contained many more structurally morphed RNAs, particularly for genes involved in the unfolded protein response and protein trafficking. “It suggests that the mechanisms leading to C9ORF72 and sporadic disease might be very different,” said Mercedes Prudencio of the Jacksonville, Florida, Mayo branch, one of the first authors on the study.
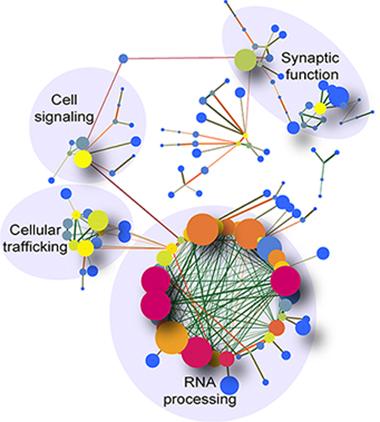
Network analysis.
RNA-processing genes in the cerebellum make up a large fraction of dysregulated RNAs in C9ORF72-linked ALS. [Courtesy of Prudencio et al., Nature Neuroscience.]
The researchers, led by joint senior authors Leonard Petrucelli in Florida and Hu Li of the Rochester, Minnesota, clinic, have put their data—encompassing RNA expression levels, alternative splicing, and alternative 3’ UTR lengths—online. Other scientists can now compare animal or iPS model changes to human brain regulation. “The public availability of these types of data sets are essential to push the field forward and will be very valuable for testing new hypotheses,” commented Adrian Isaacs of University College London, who was not involved in the study. “We will certainly be mining the data.”
Prudencio and co-first author Veronique Belzil in Florida, who designed the study, wanted to distinguish RNA processing defects caused by mutations in C9ORF72 from those caused by TDP-43, another ALS protein that warps the transcriptome. Avoiding tissues with extensive TDP-43 pathology, such as the spinal cord, they focused on the frontal cortex and cerebellum of eight C9ORF72 cases, 10 sporadic ALS cases with no known mutation, and nine neurologically healthy controls. Though C9ORF72 expansions can also cause frontotemporal dementia, Petrucelli and colleagues stuck to pure ALS in this study.
Along with joint first authors Christian Ross in Rochester and Ranjan Batra, now at the University of California in San Diego, the researchers identified the genes upregulated or downregulated in both kinds of ALS, as well as those alternatively spliced. In addition, they analyzed the presence of alternative polyadenylation sites in the pre-mRNAs. These loci indicate where RNA polymerase should stop copying DNA and add a poly-adenine tail. Alternative sites can lead to longer or shorter 3’ UTRs, which may then harbor different sites for microRNA and regulatory proteins, and alter that RNA’s regulation, Prudencio explained.
The authors hoped to find similarities between C9ORF72 and sporadic cases that would point to common mechanisms or even biomarkers. In that, they were surprised. “We identified two completely different brain profiles,” Belzil said. In C9ORF72 cases, 361 genes were differentially expressed in the cerebellum, and 241 in the frontal cortex. Expression of only 136 genes was altered in each of those regions in sporadic cases. Fifty-seven up- or downregulated cerebellum genes, and 32 in the frontal cortex, were common to both kinds of ALS.
In terms of RNA structure, alternative polyadenylation arose in 1,437 of C9ORF72 cerebellum RNAs and 716 in sporadic cerebella, and in 968 of frontal cortex RNAs of C9ORF72 cases versus 1,266 in sporadic. Alternative splicing occurred in 8,224 genes in the cerebella of C9ORF72 ALS brains, versus 2,229 in sporadic samples. In the frontal cortex, C9ORF72 brains exhibited 902 splicing changes, to 282 in sporadic cases. “It seems C9ORF72 mutations lead to worse RNA defects,” Belzil said.
The authors applied different bioinformatics tools to group the altered RNAs by function. For C9ORF72, misregulation of the unfolded protein response and protein and vesicle transport, including across the nuclear envelope, were particularly common (see genes with altered exon splicing, specifically, in image above). In contrast, alterations in sporadic cases clustered in cellular defenses and synaptic transmission. These differences suggest that treatments, and clinical trials, may need to focus specifically on one or the other ALS type, Prudencio concluded.
She and her colleagues were also surprised by the extent of RNA changes in the cerebellum. The authors wrote that despite the cerebellum’s importance for movement, scientists seem to have overlooked its involvement in ALS. However, their findings are in line with recent studies reporting cerebellar atrophy in the disease (Tan et al., 2014; Irwin et al., 2013). The cerebellum expresses more C9ORF72 than the cortex, as well as more of the C9ORF72 RNA foci and its translated repeat dipeptides, commented Edward Lee of the University of Pennsylvania Perelman School of Medicine in Philadelphia, who was not involved in the paper.
Some of the other gene networks highlighted in the study also match findings from other research models, noted Ana Jovicic of Stanford University in Palo Alto, California, who did not participate in the work. For example, ribosomal RNA (rRNA) processing genes were misregulated in both the cerebella and frontal cortices of C9ORF72 cases, and another lab recently reported crippled rRNA maturation in the blood cells and motor cortices of people who had C9ORF72-based ALS (see Haeusler et al., 2014). The dipeptides created by C9ORF72 translation also have the potential to interfere with rRNA processing (see Aug 2014 news). In addition, Petrucelli and colleagues identified misregulation of nuclear transport genes, a topic that has been getting attention in recent ALS meetings, Jovicic said.
Researchers should be able to make similar comparisons with the new database, Lee said. For example, scientists working with iPS cells in culture might zero in on a new mechanism for disease, but want to check its relevance to human ALS in the brain. “A lot of things could be checked with this data set,” he said.
Plus, this publication is just a first taste, Belzil said. The lab plans to generate a full profile of sporadic and C9ORF72 ALS brains, including epigenetic markers, in the future. They also plan to investigate the transcriptomes of blood samples, still hoping for a good biomarker, and of frontotemporal dementia cases. The group might also follow up on some of the specific genes identified in the transcriptome analysis, Prudencio said, but have not settled on any favorites yet.
In addition, researchers still have to explain how C9ORF72 creates such transcriptional havoc. Prudencio noted that the abnormal RNA foci transcribed from the repeat sequence are known partners of RNA-binding proteins like hnRNPH, and may sequester them from their normal functions (see Cooper-Knock et al., 2014; Lee et al., 2013). Supporting that theory, she and her co-authors noticed that many of the alternatively spliced genes contained an hnRNPH-binding motif.—Amber Dance
References
News Citations
Paper Citations
- Tan RH, Devenney E, Dobson-Stone C, Kwok JB, Hodges JR, Kiernan MC, Halliday GM, Hornberger M. Cerebellar integrity in the amyotrophic lateral sclerosis-frontotemporal dementia continuum. PLoS One. 2014;9(8):e105632. Epub 2014 Aug 21 PubMed.
- Irwin DJ, McMillan CT, Brettschneider J, Libon DJ, Powers J, Rascovsky K, Toledo JB, Boller A, Bekisz J, Chandrasekaran K, Wood EM, Shaw LM, Woo JH, Cook PA, Wolk DA, Arnold SE, Van Deerlin VM, McCluskey LF, Elman L, Lee VM, Trojanowski JQ, Grossman M. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry. 2013 Feb;84(2):163-9. PubMed.
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R, Rothstein JD, Wang J. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature. 2014 Mar 13;507(7491):195-200. Epub 2014 Mar 5 PubMed.
- Cooper-Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J, Hautbergue GM, Shaw PJ. Sequestration of multiple RNA recognition motif-containing proteins by C9orf72 repeat expansions. Brain. 2014 Jul;137(Pt 7):2040-51. Epub 2014 May 27 PubMed.
- Lee YB, Chen HJ, Peres JN, Gomez-Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C, Adachi Y, Sardone V, Miller JW, Smith BN, Gallo JM, Ule J, Hirth F, Rogelj B, Houart C, Shaw CE. Hexanucleotide repeats in ALS/FTD form length-dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep. 2013 Dec 12;5(5):1178-86. Epub 2013 Nov 27 PubMed.
External Citations
Further Reading
Papers
- Prudencio M, Jansen-West KR, Lee WC, Gendron TF, Zhang YJ, Xu YF, Gass J, Stuani C, Stetler C, Rademakers R, Dickson DW, Buratti E, Petrucelli L. Misregulation of human sortilin splicing leads to the generation of a nonfunctional progranulin receptor. Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21510-5. Epub 2012 Dec 10 PubMed.
- Raman R, Allen SP, Goodall EF, Kramer S, Ponger LL, Heath PR, Milo M, Hollinger HC, Walsh T, Highley JR, Olpin S, McDermott CJ, Shaw PJ, Kirby J. Gene expression signatures in motor neuron disease fibroblasts reveal dysregulation of metabolism, hypoxia-response and RNA processing functions. Neuropathol Appl Neurobiol. 2014 Apr 18; PubMed.
- Saris CG, Groen EJ, van Vught PW, van Es MA, Blauw HM, Veldink JH, van den Berg LH. Gene expression profile of SOD1-G93A mouse spinal cord, blood and muscle. Amyotroph Lateral Scler Frontotemporal Degener. 2013 Apr;14(3):190-8. Epub 2013 Jan 8 PubMed.
- Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, Shaw PJ. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS One. 2015;10(5):e0127376. Epub 2015 May 27 PubMed.
- Satoh J, Yamamoto Y, Kitano S, Takitani M, Asahina N, Kino Y. Molecular network analysis suggests a logical hypothesis for the pathological role of c9orf72 in amyotrophic lateral sclerosis/frontotemporal dementia. J Cent Nerv Syst Dis. 2014;6:69-78. Epub 2014 Aug 24 PubMed.
- Patani R, Sibley CR, Chandran S, Ule J. Using human pluripotent stem cells to study post-transcriptional mechanisms of neurodegenerative diseases. Brain Res. 2012 Jan 5; PubMed.
News
- Chicago—RNA Inclusions Offer Therapeutic Target in ALS
- Second Study Confirms Antisense Oligonucleotides Bust RNA Aggregates
- First C9ORF72 Mice Mimic Key Pathology, Behavior
- Expansion and Methylation Go Hand in Hand in C9ORF72
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- Cloak and Dagger Clusters? How C9ORF72 Repeats Kill Is Still a Mystery
- C9ORF72’s Dirty Work Done by Problem Proteins
Primary Papers
- Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza-Johnston AE, Desaro P, Bieniek KF, DeTure M, Lee WC, Biendarra SM, Davis MD, Baker MC, Perkerson RB, van Blitterswijk M, Stetler CT, Rademakers R, Link CD, Dickson DW, Boylan KB, Li H, Petrucelli L. Distinct brain transcriptome profiles in C9orf72-associated and sporadic ALS. Nat Neurosci. 2015 Aug;18(8):1175-82. Epub 2015 Jul 20 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University College London
I think this study provides an important resource for the field. The public availability of these types of data sets are essential to push the field forward and will be very valuable for testing new hypotheses—we will certainly be mining the data.
It is interesting that the authors focused on brain areas that do not show significant degeneration in ALS. The advantage of this approach is that confounds associated with gross neuronal loss and gliosis are avoided. The big question is how well these changes reflect what is happening in degenerating neurons. It will also be interesting to determine which changes are due to RNA foci and which to dipeptide repeat protein toxicity.
View all comments by Adrian IsaacsThe most striking result of this complex transcriptome analysis was the specificity of C9ORF72-ALS cases to have alterations in the unfolded protein response pathway. It was reassuring to see that many of the genes with altered splicing were the same ALS-associated genes that are involved in RNA metabolism.
I think this technique would be easily applicable to mouse research, which would allow for RNA analysis in the brain prior to death, making it easier to identify early pathways. Since the Petrucelli lab just published their C9 mouse model (Chew et al., 2015), it would not be difficult to apply these techniques to mice of varying stages of disease progression.
References:
Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang YJ, Castanedes-Casey M, Lee CW, Jansen-West K, Kurti A, Murray ME, Bieniek KF, Bauer PO, Whitelaw EC, Rousseau L, Stankowski JN, Stetler C, Daughrity LM, Perkerson EA, Desaro P, Johnston A, Overstreet K, Edbauer D, Rademakers R, Boylan KB, Dickson DW, Fryer JD, Petrucelli L. Neurodegeneration. C9ORF72 repeat expansions in mice cause TDP-43 pathology, neuronal loss, and behavioral deficits. Science. 2015 Jun 5;348(6239):1151-4. Epub 2015 May 14 PubMed.
University of Sheffield
This work establishes that dysregulation of splicing and polyadenylation occur significantly more frequently in C9ORF72 ALS than in sporadic ALS (SALS), with region-specific changes seen in the cerebellum and frontal cortex. Understanding both the similarities and differences between C9ORF72 ALS and SALS is essential for development of therapeutic strategies that target either or both groups of patients.
It would be interesting to know how this aberrant RNA processing of the cerebellum compares to patients with C9ORF72 frontotemporal dementia, as well as whether this happens in the motor neurons and/or glial cells of the spinal cord. The dysregulation observed in the cerebellum and frontal cortex of C9ORF72 is also consistent with a recently published paper reporting aberrant RNA splicing in peripheral tissues of C9orf72-related ALS (Cooper-Knock et al., 2015). Thus, disruption of RNA processing is emerging as a major pathological mechanism in C9ORF72-ALS.
References:
Cooper-Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J, Shaw PJ. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS One. 2015;10(5):e0127376. Epub 2015 May 27 PubMed.
Make a Comment
To make a comment you must login or register.