In C9ORF72 Expansion Carriers, Protein Levels Drop in Cerebellum
Quick Links
Hexanucleotide expansions in the C9ORF72 gene are the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), yet the physiological function of the protein remains a mystery. To address this gap, researchers developed a new suite of monoclonal antibodies specific to the C9ORF72 protein, and reported their initial findings in the August 3 Acta Neuropathologica. Led by Manuela Neumann of the German Center for Neurodegenerative Diseases in Tübingen, the study found that in mouse and human neurons, C9ORF72 mingled with lysosomes, other vesicles, and even resided in synapses, where it bound Rab3 proteins that release synaptic vesicles. What’s more, expansion carriers had 20 percent less of the protein in the cerebellum than did noncarriers, contrasting previous reports that found no difference despite a loss of C9 mRNA in carriers. The researchers contend that the findings support the possibility that a dearth of C9ORF72 protein contributes to disease pathogenesis.
- Researchers raised new monoclonal antibodies to wild-type C9ORF72.
- They detect the protein in presynapses, lysosomes, and other vesicles.
- Carriers of C9ORF72 repeat expansions had 20 percent less C9ORF72 in the cerebellum.
"The development of novel and specific C9ORF72 antibodies as reagents that allow for the further characterization of the protein’s endogenous function is important,” noted Brian Freibaum, St. Jude Children’s Research Hospital, Memphis, Tennessee.
Multiple, nonexclusive hypotheses exist to explain how C9ORF72 expansions cause disease. These include two gain-of-function mechanisms, in which RNA transcribed from the expansions, or the dipeptide repeats translated from that RNA, form toxic aggregates that sequester other RNAs and proteins. Some evidence also supports the idea that loss of C9ORF72 function could play a role, since the repeat expansions reportedly hobble transcription of the gene (Feb 2018 news; Waite et al., 2014; van Blitterswijk et al., 2015). However, the physiological function of the wild-type C9ORF72 remains a mystery, and detecting it has been hampered by a lack of specific monoclonal antibodies. Neumann told Alzforum that commercially available antibodies only detect the protein when it is highly overexpressed, rendering efforts to measure physiological levels futile. She added that compared with other proteins, C9ORF72 happens to have very low antigenicity, presenting a high hurdle for antibody development.
First author Petra Frick and colleagues stepped up to the challenge. After screening more than 100 potential antibody clones raised against synthetic C9ORF72 peptides that were conjugated to albumin, they narrowed the choices down to a handful of hopefuls. The two monoclonals they settled on—dubbed 1C1 and 12E7—recognized exogenously expressed C9ORF72 in HEK293 cells, and also detected the protein at endogenous levels in the mouse CNS. Notably, the antibodies did not detect signals in C9ORF72 knockout mice, suggesting the antibodies are specific to the C9 protein. C9ORF72 exists in long and short isoforms, and the researchers found that while the antibodies bound both, they only detected the longer 481-amino acid isoform in western blot from mouse brain extracts, suggesting this was the predominantly expressed isoform in the CNS.
To see where C9ORF72 localized in neurons, the researchers used the antibodies to probe subcellular fractions of mouse brain, and also stained brain tissue sections by immunohistochemistry. They found that C9ORF72 primarily resided in the cytoplasm. The protein also appeared in synaptosomal fractions, suggesting a penchant for presynapses. Immunohistochemistry supported this idea; the researchers observed punctate immunoreactivity in neuropil regions that was consistent with synaptic localization. This synaptic staining pattern was most distinct in the mossy fibers emanating from neurons in the hippocampus, and also appeared in motor neurons in the spinal cord. In large motor neurons, the researchers also spotted C9ORF72 mingling with small vesicles in the cytoplasm. These vesicles were devoid of lysosomal or synaptic vesicle markers, and so far their identity is unclear.
What about in human samples? Unfortunately, the antibodies did not detect C9ORF72 in postmortem brain tissue samples. This was most likely due to sensitivity of the antibody epitopes to formalin fixation, the researchers proposed. In support of this idea, the antibodies also failed to detect C9ORF72 in mouse tissue after two days of formalin fixation. Neumann told Alzforum that the antibodies could work on postmortem brain tissue fixed for less than 24 hours, and that the researchers are seeking out those samples as they become available.
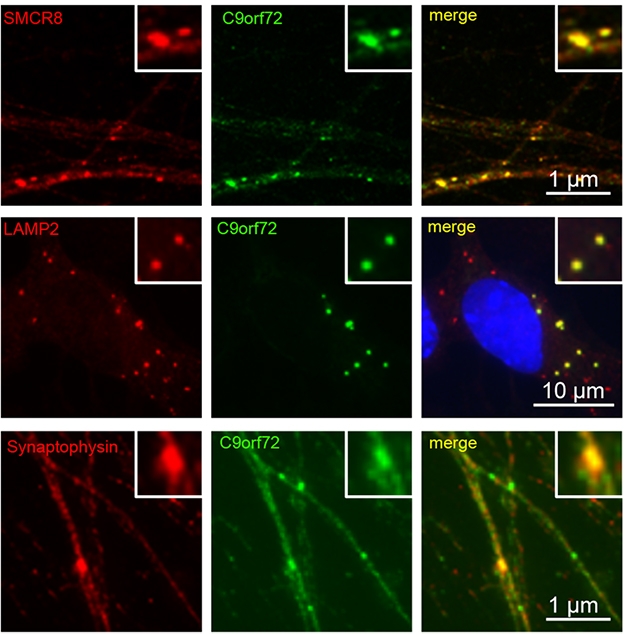
C9’s Associates. In iPSC-derived motor neurons, C9ORF72 (green) co-localized with SMCR8 (top), the lysosomal marker LAMP2 (middle), and the synaptic marker synaptophysin (bottom). [Courtesy of Frick et al., Acta Neuropathologica, 2018.]
For now, Frick and colleagues explored C9ORF72 localization in human motor neurons derived from induced pluripotent stem cells (iPSCs). The researchers spotted C9ORF72 in cytoplasmic puncta, a fraction of which co-localized with lysosomal markers LAMP1 and LAMP2. Around 11 percent of the puncta also merged with synaptic markers. C9ORF72 strongly co-localized with SMCR8, a protein previously reported to interact with C9ORF72, forming a complex that acts as a guanine nucleotide exchange factor. This GEF activated Rab proteins involved in autophagy, according to those studies (Sellier et al., 2016; Sullivan et al., 2016). Frick found that C9ORF72 co-localized with members of a different Rab protein family, Rab3, which facilitates release of synaptic vesicles into the active zone of the synapse (Binotti et al., 2016). While the role of C9ORF72 in this process remains unclear, Neumann speculated that C9ORF72 could help Rab3 proteins release vesicles.
Finally, the researchers wanted to know whether the presence of hexanucleotide expansions in C9ORF72 affected expression of the protein. To test this, they acquired freshly frozen cerebellar tissue from 18 expansion carriers with ALS, ALS/FTD, or FTD, as well as 33 noncarriers, who had been neurologically healthy, had Alzheimer’s disease, or had non-C9ORF72-related type ALS or FTD. Because the cerebellum does not degenerate in ALS/FTD, the researchers reasoned that measuring C9ORF72 protein there would avoid confounds of neuronal loss in more vulnerable regions, such as the frontal cortex or spinal cord. They found that compared with noncarriers, expansion carriers had about 20 percent less of the protein in the cerebellum. However, the cerebellar C9ORF72 concentrations did not correlate with the clinical phenotype of the carriers. Nevertheless, Freibaum thought that the loss might be important. “Though the 20 percent reduction in protein quantity seems small, this could have dramatic effects over the lifespan of the neurons, especially coupled with gain-of-function mechanisms or impairment of autophagy,” he wrote.
The cerebellum data contradicts some previous reports, which found no reduction in protein levels in the region in expansion carriers, despite less mRNA there, even as the protein plummeted in the cortex (Waite et al., 2014; Aug 2015 news). The researchers attributed these differences to the higher sensitivity of their antibodies, claiming that the drop in C9ORF72 protein levels in degenerating regions of the brain may have been easier to pick up with less sensitive antibodies. Neumann is currently sharing the new antibodies with interested collaborators, and told Alzforum she has initiated the process of making them available commercially as well.—Jessica Shugart
References
News Citations
- Lack of C9ORF72 Protein Renders Neurons More Vulnerable to Degeneration
- New C9ORF72 Antibodies Find Isoforms in Different Cellular Locations
Paper Citations
- Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014 Jul;35(7):1779.e5-1779.e13. Epub 2014 Jan 17 PubMed.
- van Blitterswijk M, Gendron TF, Baker MC, DeJesus-Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Petrucelli L, Boeve BF, Graff-Radford NR, Boylan KB, Dickson DW, Rademakers R. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015 Dec;130(6):863-76. Epub 2015 Oct 5 PubMed.
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet-Berguerand N. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016 Jun 15;35(12):1276-97. Epub 2016 Apr 21 PubMed.
- Sullivan PM, Zhou X, Robins AM, Paushter DH, Kim D, Smolka MB, Hu F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol Commun. 2016 May 18;4(1):51. PubMed.
- Binotti B, Jahn R, Chua JJ. Functions of Rab Proteins at Presynaptic Sites. Cells. 2016 Feb 6;5(1) PubMed.
Further Reading
Primary Papers
- Frick P, Sellier C, Mackenzie IR, Cheng CY, Tahraoui-Bories J, Martinat C, Pasterkamp RJ, Prudlo J, Edbauer D, Oulad-Abdelghani M, Feederle R, Charlet-Berguerand N, Neumann M. Novel antibodies reveal presynaptic localization of C9orf72 protein and reduced protein levels in C9orf72 mutation carriers. Acta Neuropathol Commun. 2018 Aug 3;6(1):72. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Cardiff University
Since the discovery of the C9ORF72 hexanucleotide repeat expansion as a causative mutation in ALS-FTD, a number of studies have used various cellular and animal models to explore the normal cellular roles of C9ORF72 protein isoforms and to determine if and how haploinsufficiency of C9ORF72 contributes to disease pathogenesis. However, studies of endogenous C9ORF72 protein isoforms have been hampered by the lack of commercial antibodies with high specificity and sensitivity. A small number of antibodies have also been developed by academic groups, including our group (Waite et al., 2014; Koppers et al., 2015), but there is still a lack of knockout validated C9ORF72 antibodies that are suitable for biochemical, immunocytochemical, and immunohistochemical studies. The study reported here describes the generation, characterization, and preliminary use of novel mouse and rat anti-C9ORF72 monoclonal antibodies generated against both long and short protein isoforms. The authors performed detailed characterization of the monoclonal antibodies using analysis of biochemical, immunocytochemical, and immunohistochemical methods including the use of C9ORF72 knockout mouse tissue to verify specificity. A parallel characterization of commercially available antibodies demonstrated their lack of specificity in biochemical and immunohistochemical analyses (in agreement with our published study of commercial antibodies using siRNA-mediated knockdown of C9ORF72 in a human cell line (Waite et al., 2014). They used their own validated mouse 1C1 and rat 12E7 antibodies (that can detect both long and short C9ORF72 protein isoforms) in further analyses of mouse tissue, hiPSC-derived motor neurons and human postmortem tissue.
The authors used the 1C1 and 12E7 monoclonal antibodies to demonstrate that the long C9ORF72 protein isoform is predominantly detected in mouse and human tissue by western blot (in agreement with our own findings in human cells and tissue (Waite et al., 2014) with multiple tissue western blot analysis showing highest levels of protein detected in the CNS and spleen. Immunohistochemistry analysis of mouse tissue using a knockout validated protocol revealed a punctate neuropil staining pattern throughout the CNS reminiscent of synaptic localisation (showing an overlapping immunostaining pattern with the presynaptic marker synaptoporin) with highest levels of reactivity in the hippocampal mossy fiber system and globus pallidus, with weaker signals in the neuropil of the cortex, caudate-putamen, and molecular layer of the cerebellum. C9ORF72 was also localised to cytoplasmic punctae (identity unknown) in the cell bodies of large motor neurons in the brain stem and spinal cord. No immunoreactivity was detected in white matter or glial cells. Subcellular fractionation experiments also demonstrated the cytosolic localisation of C9ORF72 with an enrichment in synaptosomes (enriched in synaptic vesicles).
Extending the study to immunocytochemical analysis of hiPSC-derived motor neurons demonstrated a punctate-staining pattern with high co-localization with the SMCR8 protein, a robust interaction partner of C9ORF72 (Sellier et al., 2016), and partial co-localization with the lysosomal marker LAMP2, in agreement with previous studies (Amick et al., 2016) and the synaptic vesicle marker synaptophysin. Furthermore, the authors demonstrate an association of C9ORF72 with the RAB3 protein family of synaptic vesicle localized GTPases, which are essential for normal neurotransmitter release, via co-immunoprecipitation of recombinant and endogenous material, and they found evidence of partial co-localization in hiPSC-derived motor neurons. Therefore, several lines of evidence suggest that a pool of C9ORF72 may be involved in synaptic vesicle function, however, this requires further mechanistic studies in animal and cellular models, as noted by the authors.
Finally, the authors use their validated antibodies to analyze C9ORF72 protein levels by western blot of postmortem brain tissue of C9ORF72 mutation cases and neurologic controls. They demonstrate that decreasing C9ORF72 protein levels correlate with general neuronal loss thus, complicating the analysis of tissues undergoing neurodegeneration as shown in frontal cortex samples from a neurodegeneration panel (AD, ALS, FTD, and hypoxia). As an alternative, they perform western blot analyses of cerebellum tissue that escapes overt neurodegeneration in ALS-FTD. C9ORF72 showed a reduction of approximately 20 percent in hexanucleotide repeat expansion cases versus controls and displayed no alteration in protein solubility. In contrast to western blotting, the antibodies could not be validated for use in immunohistochemical analysis of postmortem brain tissue. The authors propose that this could be due to the extended formalin fixation times for human tissue versus mouse tissue. Further studies of human postmortem tissue will require modified fixation conditions for improved detection of C9ORF72 using immunohistochemistry.
This study provides additional support for reduced C9ORF72 protein as a consequence of the hexanucleotide repeat expansion, however it remains to be conclusively determined if and how reduced protein levels contribute to disease pathogenesis. Published studies of human ALS-FTD postmortem material, and C9ORF72 and Smcr8 deficient mouse models (Koppers et al., 2015; Sudria-Lopez et al., 2016; Atanasio et al., 2016; Burberry et al., 2016; Zhang et al., 2018) suggest that haploinsufficiency is insufficient to cause ALS-FTD. However, it is possible that a combination of reduced C9ORF72 function, hexanucleotide repeat-associated gain-of-function mechanisms, and other cellular stressors ultimately lead to disease progression and may explain the phenotypic heterogeneity associated with the C9ORF72 repeat expansion (see Shi et al., 2018, and recent review in Balendra and Isaacs, 2018). The availability of knockout-validated monoclonal antibodies provides an abundant resource for future studies of clinical material, to study cellular and animal models aimed to further investigate the context-dependent expression, for the localization and function of C9ORF72, and to elucidate the contribution of loss-of-function to ALS-FTD pathogenesis.
References:
Amick J, Roczniak-Ferguson A, Ferguson SM. C9orf72 binds SMCR8, localizes to lysosomes, and regulates mTORC1 signaling. Mol Biol Cell. 2016 Oct 15;27(20):3040-3051. Epub 2016 Aug 24 PubMed.
Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y, Fury W, Burfeind P, Zamfirova R, Warshaw G, Orengo J, Oyejide A, Fralish M, Auerbach W, Poueymirou W, Freudenberg J, Gong G, Zambrowicz B, Valenzuela D, Yancopoulos G, Murphy A, Thurston G, Lai KM. C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep. 2016 Mar 16;6:23204. PubMed.
Balendra R, Isaacs AM. C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol. 2018 Sep;14(9):544-558. PubMed.
Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki-Uematsu S, Ghosh S, Singh A, Merkle FT, Koszka K, Li QZ, Zon L, Rossi DJ, Trowbridge JJ, Notarangelo LD, Eggan K. Loss-of-function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med. 2016 Jul 13;8(347):347ra93. PubMed.
Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH, van den Berg LH, Pasterkamp RJ. C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol. 2015 Sep;78(3):426-38. Epub 2015 Jul 3 PubMed.
Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb-Cheynel I, Oulad-Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E, Charlet-Berguerand N. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016 Jun 15;35(12):1276-97. Epub 2016 Apr 21 PubMed.
Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY, Wen X, Kisler K, Wilkinson B, Menendez L, Sugawara T, Woolwine P, Huang M, Cowan MJ, Ge B, Koutsodendris N, Sandor KP, Komberg J, Vangoor VR, Senthilkumar K, Hennes V, Seah C, Nelson AR, Cheng TY, Lee SJ, August PR, Chen JA, Wisniewski N, Hanson-Smith V, Belgard TG, Zhang A, Coba M, Grunseich C, Ward ME, van den Berg LH, Pasterkamp RJ, Trotti D, Zlokovic BV, Ichida JK. Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med. 2018 Mar;24(3):313-325. Epub 2018 Feb 5 PubMed.
Sudria-Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, Youssef SA, Harkema L, de Bruin A, Veldink JH, van den Berg LH, Pasterkamp RJ. Full ablation of C9orf72 in mice causes immune system-related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol. 2016 Jul;132(1):145-7. Epub 2016 May 20 PubMed.
Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging. 2014 Jul;35(7):1779.e5-1779.e13. Epub 2014 Jan 17 PubMed.
Zhang Y, Burberry A, Wang JY, Sandoe J, Ghosh S, Udeshi ND, Svinkina T, Mordes DA, Mok J, Charlton M, Li QZ, Carr SA, Eggan K. The C9orf72-interacting protein Smcr8 is a negative regulator of autoimmunity and lysosomal exocytosis. Genes Dev. 2018 Jul 1;32(13-14):929-943. Epub 2018 Jun 27 PubMed.
Make a Comment
To make a comment you must login or register.