Begone 2020: Despite COVID, Alzheimer’s Research Advanced
Quick Links
Last year was a year to remember. (Or make that a year to forget!) Hope held until February that SARS-CoV-2 might be contained, but was quickly dashed when the virus spread across the globe. Lockdowns, social isolation, protective equipment shortages, and heartbreak became the norm as millions of people became infected and the death toll soared, especially among the old.
People with dementia were most vulnerable, and this constrained research. Clinical studies and trials were hampered because in-person visits had to be cancelled or rescheduled (Mar news; Mar news). Basic research was disrupted. Laboratories worldwide were forced to shutter or curtail access, putting projects on ice and precious cell and animal lines in jeopardy (see image below and Mar news). Conference organizers scrambled to reschedule or switch to an online format, with various degrees of success (Oct news). Casualties included the Society for Neuroscience annual meeting. Typically drawing up to 30,000 scientists annually, it was cancelled.
By June, investigators had adjusted and research began to get mostly back on track (Jun news). Some clinical trials were abandoned (Jun news), but even so, as outlined in this yearly research roundup, the field overall managed to make strides in important areas, from plasma tests for Aβ and tau to therapeutics. Twenty-twenty saw the first—if embattled—FDA filing for a new AD drug since 2003. If approved, it would be the first biologic treatment for this disease.

Lonesome in the Lab. At DZNE’s Biomedical Research Center near Munich, white window shades open automatically when rooms are occupied (left). On March 25, a Wednesday, only two people, the director and a technician, were there. [Courtesy of Christian Haass.]
Therapeutics
The drama of aducanumab was the year’s big story. Following on 2019’s head-spinning plot turns of futility stoppage, resurrection, and post-hoc salvage partnership with the FDA, 2020 proved to be no less of a page-turner.
In summer, Biogen applied for marketing approval with the U.S. FDA (Jul news), in October with Europe’s EMA, and in December with Japan’s Ministry of Health, Labor, and Welfare. Alas, in November, days before the FDA’s expert advisory committee hearing, the agency posted public briefing materials that sparked consternation among scientists. Critics detected an unusually close relationship between the drug’s sponsor and its regulating agency, and charged that the data of the two incomplete and contradictory trials did not prove aducanumab worked. During the hearing, Biogen and FDA neurologists endured blistering criticism from independent advisers, even as patient advocates offered efficacy anecdotes and repeated the known pleas about the desperate medical need of sufferers and their families (Nov news). Opinion in the field is split between wanting data from a confirmatory trial and wanting to approve now, warts and all (Nov news).
On December 9, in the year’s final twist, the Washington, D.C.-based advocacy group Public Citizen demanded that FDA Commissioner Stephen Hahn reassign the review of Biogen’s licensing application. Public Citizen also requested an investigation from the U.S. Department of Health and Human Services’ Office of Inspector General (letter to FDA; letter to the Inspector General). Such is the climate in which the FDA now deliberates. A decision is expected by March 5.
Some readers may not have noticed, but a sponsor besides Biogen is awaiting the FDA’s thumbs up or down by this spring. In June 2020, Acadia Pharmaceuticals, the maker of the anti-psychotic pimavanserin, submitted a marketing application to treat dementia-related psychosis.
Overshadowed as they were by aducanumab, other anti-amyloid drugs made news last year. Eisai’s BAN2401, now called lecanemab, began enrolling for AHEAD 3-45, a 1,400-participant-strong, four-year-long secondary prevention trial run by the Alzheimer's Clinical Trial Consortium (ACTC) (Nov conference news). An ambitious screening campaign is underway. Separately, TRC-PAD—the long-planned trial-ready cohort for preclinical AD trials more generally—has emerged from COVID delays and is finally up and running, as well (see image below and Nov conference news).

Into the Funnel. Thousands of participants take online cognitive tests in the APT Webstudy. Those who show hints of cognitive decline are referred to nearby study sites (SRS) for in-clinic tests, selection for amyloid screens, and, if positive, inclusion in the trial-ready cohort (TRC).
In the fall, Roche started infusing participants with gantenerumab in an open-label extension of the first DIAN-TU cohort trial. This antibody had fallen short of the trial’s primary outcome, but it did push amyloid, tau, and the NfL neurodegeneration biomarker in the desired direction (Apr conference news). Participants tolerated gantenerumab well, hence Roche is now evaluating higher doses in a separate trial of weekly injections under the skin. Otherwise, 2020 was quiet for gantenerumab, as its two large, multiyear Phase 3 trials navigated the pandemic toward their 2022 readout. Eli Lilly & Co began a 500-person, Phase 2 trial of the pyroglutamate Aβ antibody donanemab in 2020.
Beyond therapeutic antibodies, there was some action on the anti-amyloid front. Twenty-twenty saw the start of Phase 3 trials for the candidate vaccine UB-311, the Alzhemed remake ALZ-801, and the approved-in-China oligosaccharide GV-971. Defeat came in Phase 2/3 for azeliragon and the green tea extract EGCG. In contrast, the battle continues for the sigma2/PGRMC1 receptor antagonist Elayta, which received the green light for a 540-person ACTC trial. Ditto for varoglutamstat, formerly known as PQ912. In 2020, a 250-participant Phase 2 trial of this glutaminyl cyclase inhibitor began in Europe, and a larger U.S. trial with the Alzheimer Disease Cooperative Study will start soon.
Finally, BACE inhibitors. Their trials were waylaid before 2020 by a detrimental class effect on cognition. It might seem that nothing would snuff out a candidate Alzheimer’s drug like hurting cognition. And yet, BACE inhibitors are not quite dead. Throughout 2020, pharma and academic scientists have, sometimes collaboratively, scrutinized the trial data to learn what went wrong. The upshot? It seems all those trials might have given far too high a dose. Even so, the drugs may not have hastened AD neurodegeneration and, in cultured neurons, low doses preserve synaptic function (Dec news; Jun news). Do these drugs deserve a second chance? Pharma has shown no appetite for low-dose trials thus far, and is looking toward public or public-private initiatives.
The other big target in 2020—tau—continued to baffle scientists with its astounding complexity in the form of post-translational modifications (see image below and Dec news).

Post-translational Tau. Different modifications bedeck the length of the tau protein. Tau proteoforms also differ based on how many N-terminal inserts they contain (0N, 1N, or 2N) and whether they contain three or four microtubule-binding domains (3R or 4R). [Courtesy of Wesseling et al., Cell.]
Consequently trialists, too, are realizing that tau is not a monolithic target. They now believe it may take many shots at different forms of tau until they land a hit. Already in 2019, the first tau-targeted antibodies had started failing in the clinic, and in 2020 this disquieting trend continued when Genentech’s semorinemab gave no efficacy signal whatsoever in the first of its two Phase 2 Alzheimer’s trials. Abbvie’s tilavonemab and Biogen’s gosuranemab posted negative results in progressive supranuclear palsy, though their respective Alzheimer’s trials are still ongoing.
Ouch. Curiously, though, all three antibodies bind epitopes at tau’s N-terminus. Hence, the field’s interest has shifted toward tau’s middle section, prompting a flurry of attacks on a proline-rich domain there, as well as the adjacent microtubule binding domain. Lilly’s zagotenemab entered Phase 2 in 2020. So did JNJ-63733657. Nipping at their heels are UCB’s bepranemab, Biogen’s BIIB076, Eisai’s E2814, Lundbeck’s LU-AF87908, and Pinteon’s PIN001.
The field’s two active tau vaccines made incremental progress. ADDvac1 posted Phase 2 biomarker data, and its sponsor, Axon Neuroscience, plans a Phase 3 this year. AC Immune’s original ACI-35 vaccine proved to be insufficiently immunogenic and a stronger backup, ACI-35.030, has entered Phase 1. So did small-molecule inhibitors of tau oligomer aggregation and of the tau-modifying O-GlcNAcase enzyme, as well as an antisense oligonucleotide (Apr conference news; Therapeutics database).
Alzheimer’s disease pathogenesis encompasses more than plaques and tangles, hence drug development has branched out well beyond this central pathway. The past decade of basic research on mechanisms of inflammation has started to yield new targets, and translational science is now feeding compounds into trials. In 2020, the Trem2 antibody AL002 appeared safe and able to reach the CSF, and entered Phase 2. A small molecule inhibiting another hot-button target, the NLRP3-containing inflammasome, posted good safety data in March in a first-in-human study, and is alleged to enter the brain. Called inzomelid, its developer Inflazome was promptly swallowed up by Roche for future development of inzolemid in neurodegenerative diseases (September C&EN News). Pepinemab, an anti-semaphorin 4D antibody meant to keep glia from spewing certain cytokines, posted promising data in Huntington’s and last fall entered its first Alzheimer’s trial.
An older anti-inflammatory compound gained momentum in 2020. The repurposed p38 MAPK kinase inhibitor neflamapimod, which had started out as an arthritis drug, has been in early stage AD trials for some years. Last November, its sponsor reported that a small Phase 2 trial in dementia with Lewy bodies met its primary endpoint.
Besides trying to steer glial activation away from neurotoxic cytokine release and toward phagocytosis, scientists are also hoping to prop up healthy proteostasis in aging neurons to avoid misfolded protein aggregation in the first place. One such effort inched forward in 2020. DNL343 is a small molecule that tamps down the unfolded protein response by ratcheting up the protein translation initiator EIF2b. It entered Phase 1 with a first trial in February and a second in December. An epigenetic approach, attempting to slow histone demethylation by way of the small molecule vafidemstat appeared to have safely nudged inflammation and neurodegeneration biomarkers in Phase 2a.
For Lewy body disease and Parkinson’s therapies—some of which may prove useful for AD—the year 2020 was mixed. α-Synuclein antibodies hit troubled waters, with Roche’s prasinezumab missing its primary endpoint in Phase 2, and a comparative study of putative conformation-specific antibodies judging them all to be insufficiently discerning (Apr conference news; Oct news). But therapy research in this field has branched out, too, and some tender shoots show promise. For example, a LRRK2 inhibitor is finishing Phase 1, and a GBA-activating small molecule as well as a Toll-like receptor 2 inhibitor to boost autophagy both hit their targets in Phase 1 (DNL201, LTI-291, NPT520-34).
Fluid Markers
Precivity, C2N’s mass spectrometry-based test to measure the Aβ42/40 ratio, took a leap forward in October. The St. Louis-based company won CLIA approval for its plasma test in the U.S. and the EU. Cheaper than a PET scan, the test could help clinicians diagnose AD. Hot on Precivity’s heels are immunoassays for various phosphorylated forms of tau that spill into the plasma early in the disease (Nov news). This field is moving so fast, it’s hard to keep track. In March, new data bolstered the idea that plasma p-tau181 levels correlate with plaques and tangles, distinguish people with AD from controls and other tauopathies, and predict cognitive decline (Mar news). In July, plasma p-tau217 started looking even better, outperforming plasma p-tau181, plasma neurofilament light (NfL), and even imaging markers (see image below and Jul conference news).

Tangles versus p-Tau. As amyloid burden grows in the brain as seen by PET (x axis), plasma and CSF p-tau217 changes outpace tangle accumulation. [Courtesy of Janelidze et al., JAMA Neurology 2020.]
As do plasma Aβ42 and p-tau, plasma NfL begins to change well before symptom onset, as many as 22 years prior in people with familial AD mutations (May news). NfL is an unspecific marker of neurodegeneration that may help clinicians stage the disease, monitor treatments in trials, and generally round out a toolbox of diverse fluid markers scientists are assembling.
Other tau fragments are joining the mix. In November, plasma levels of NT1, an N-terminal fragment, were shown to predict tangle accumulation, cognitive decline, and neurodegeneration, while December saw reports of a microtubule binding domain fragment in the CSF that tracked with tau PET and dementia (see image below and Dec news; Dec news). Earlier in the year, p-tau181/p-tau-217, p-tau205, and total tau in the CSF were linked to early amyloidosis, onset of brain shrinkage, and the beginning of tau tangles, respectively (Mar news).

Forecasting Neurodegeneration. Higher NT1 at baseline means greater longitudinal neurodegeneration as seen by gray-matter volume (left), hippocampal volume (center), and cortical thinning (right), particularly for people with a high plaque burden (solid lines in a and b).
Last August, the field lost a pioneer of tau research, Peter Davies, 72 (Sep news). Many of the antibodies and animal models Davies developed remain in use today.
Cryo-EM
Once again this year, single molecule cryo-electron microscopy and tissue-level cryo-electron tomography dazzled eyes and minds. Tau fibrils from people with corticobasal syndrome were found to be distinct from those in AD, Pick’s disease, and chronic traumatic encephalopathy, supporting the idea that different fibrils arise from different “strains” of the same protein (Feb news).
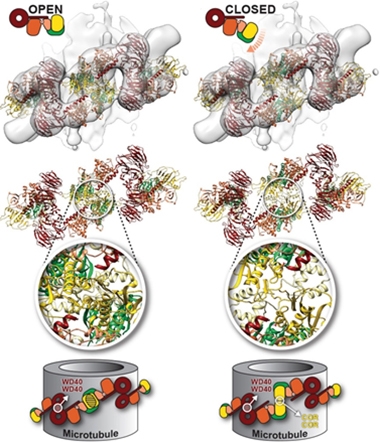
Square Peg, Round Hole. In the open-kinase conformation (left), the LRRK2 monomer fits poorly into the filament structure. With its kinase (orange) closed (right), it clicks into place. [Courtesy of Deniston et al., Nature.]
Cryo-EM revealed the first structures of α-synuclein fibrils, extracted from the brains of people with multiple system atrophy. These fibrils were nothing like previous ones made in vitro, and were distinct from Aβ or tau fibrils (Mar conference news; May news).
Twenty-twenty saw the first native structures of LRRK2 (see image at left). Cryo-electron tomography of kidney-cell sections captured whole filaments of this massive kinase wrapped as double helices around microtubules, revealing open and closed conformations. Cryo-EM of a fragment of the protein to a resolution of 3.5Å fit the double-helix structure, but only if the kinase was in its closed, catalytically active conformation (Aug news).
In October, two groups independently announced how better hardware and software had enabled them to break the atomic-resolution barrier using cryo-EM. They determined the shape of a benchmark protein for structural studies down to 1.2Å, rivaling X-ray crystallography (Oct news). One group solved the structure of the pentameric human GABAA receptor to 1.7Å. Other formerly recalcitrant structures, such as those of membrane proteins, will soon follow.
Brain Imaging
In 2020, the young field of imaging neurofibrillary tangles in the living brain matured a bit. The new PET tracers JNJ-067 and SNFT-1 appear to bind more specifically to tangles in AD tissue than does flortaucipir; aka Tauvid, which received FDA approval in May. MK-6240 emerged as perhaps the most sensitive ligand; and evidence tied the tracers PI-2620 and APN-1607 to 4R tau, meaning they detect tangle pathology in primary tauopathies such as progressive supranuclear palsy and corticobasal degeneration (see image below and Feb conference news; Mar conference news; and Jul news). Comprising only 4R tau, those tangles are structurally different from the 3R/4R hybrids that form in AD. In contrast, Roche’s tracer RO-498 proved highly specific for those, even outperforming amyloid PET and CSF Aβ in distinguishing AD from other dementias (May news).

Inclusive Tracer? PI-2620 binds to 4R aggregates in the tauopathies PSP (second row) and CBD (third row), and to paired helical filaments of tau in AD (bottom row), while having little background in healthy controls (top row). [Courtesy of Roesler et al., Progress in Neurobiology.]
Researchers reported that neurofibrillary tangles in AD appear first in the rhinal sulcus, that connectivity best predicts their spread, and that tau PET predicts brain atrophy (see image below; Mar conference news; Jan news; May news; Jan news). An individualized model of tau spread based on a person’s connectivity could reduce the number of people needed in tau therapy trials by up to two-thirds, researchers claimed (Nov news). Other work further bolstered the old idea that tau pathology only takes off after amyloid has carpeted the cortex (Feb conference news).
On the amyloid PET front, reimbursement hopes took a hit when the IDEAS study found that a PET scan reduced hospitalizations the following year by only 5 percent, falling short of the prespecified endpoint of 10 percent that would have suggested a cost benefit (Aug conference news).
In other PET news, the synaptic vesicle tracer UCB-J detected fewer synapses throughout the cortex of people with AD and other neurodegenerative diseases, and inversely correlated with tangles and cognitive decline (Feb conference news; Feb conference news).
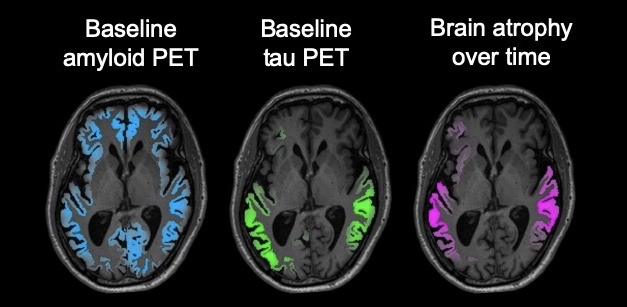
Atrophy Follows Tangles. In a person with early AD, a tau PET scan (green) predicts where the brain will shrink (magenta) better than an amyloid scan (blue). [Courtesy of the Rabinovici lab/UCSF.]
Glia
Rodents only go so far in modelling human disease, and nowhere is that more apparent than in trying to decipher the role of microglia in neurodegeneration. Single-nucleus RNA sequencing (snRNA-Seq) has enabled scientists to tease out in one fell swoop which genes change expression among thousands of individual brain cells. In this way, they had previously found that mouse and human microglia switch on different transcriptomes when tackling amyloidosis (May 2019 news).
Support for this idea came in January 2020, with the finding that while responses of both mouse and human glia to amyloid depended on TREM2 signaling, mouse glia turned down homeostatic genes while human microglia turned them up (Jan news). This was not the end of the story. In October, snRNA-Seq’s suitability for studying human frozen tissue came into question because it misses transcriptional changes in AD microglia that manifest in the cytoplasm (see image below and Oct news).
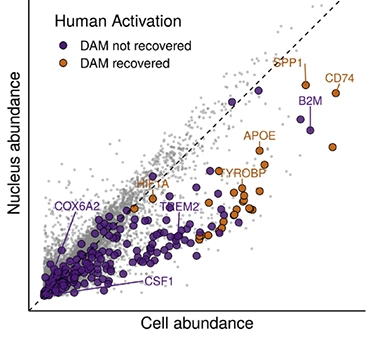
DAMN Scarce. Because disease-associated microglial (DAM) transcripts are in short supply in the nucleus, only the most abundant transcripts (gold) can be detected in nuclei from activated human microglia, while scarcer ones (purple) are missed. [Courtesy of Thrupp et al., Cell Reports.]
Clarity may come from single-cell RNA-Seq to capture transcriptomes of live microglia that were freshly isolated from human brains immediately after death or from a tissue biopsy. These tissues are hard to come by but, in 2020, scientists at a growing number of centers worked with surgery departments to obtain them. One such study bolstered the idea that human microglia are mostly homeostatic, even in disease, and that no one subgroup expresses disease-associated microglial genes found in mice. Instead, the mouse DAM genes are active in all of nine human microglial subtypes identified by transcriptome clustering (Dec news).
A given glial cell’s response appears to depend on how close it is to trouble. A study marrying spatial transcriptomics with in-situ sequencing identified gene activation changes that only occurred adjacent to plaques, in both mouse and human brains. Microglia and oligodendrocytes changed the most (Jul news).
Not only where, but also when genes turn on an off is undoubtedly important. Toward a better grasp of this, 2020 saw a way to “timestamp” RNA, which could help scientists determine the temporal sequence of gene activation in response to a stimulus (see image below and Oct news).
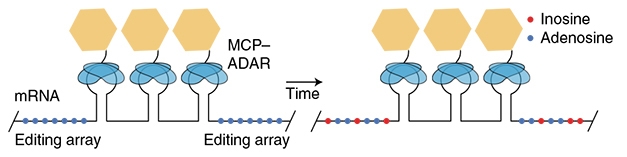
Timing mRNA. A tag added to messenger RNA includes three MS2 binding sites (keyhole shapes) amid adenosine residues (blue dots). The MS2 domains recruit a modified ADAR enzyme (yellow and blue), which gradually converts adenosines to inosines (red). [Courtesy of Rodriques et al., Nature Biotechnology.]
How glia behave in pathogenesis has become one of the most active areas of AD research. A massive proteomics study of more than 2,000 AD and control brain samples identified modular changes among more than 3,000 proteins that suggested both microglia and oligodendrocytes rev up their metabolism early (May news). Scientists now generally believe that in AD and its mouse models, microglia nibble away at synapses, contributing to neurodegeneration and memory loss. In July, researchers proposed that the neuronal protein SRPX2 might deter microglia from pruning too much (Jul news). SRPX2 steers complement factor C1q away from synapses, where it acts as an “eat me” signal for microglia. On the other hand, such synaptic remodeling might be essential for normal learning and memory. By clearing the extracellular matrix near synapses, microglia lay the groundwork for new ones, helping the brain form new memories and maintain old ones (Jul news).
What happens when neurons start dying? Once things are this dire, glia collaborate to clear up the debris. When scientists used two-photon apoptotic targeted ablation to kill off individual neurons in living mice, they found that astrocytes gobbled up distal dendrites while microglia went for the proximal dendrites and the soma in a coordinated fashion (see image below and Jun news).

Division of Labor. In mouse brain, microglia (green) clean up the soma and proximal dendrites of a dying neuron (white); astrocytes (red) mop up distant dendrites. Where green meets red, there is a boundary. [Courtesy of Damisah et al., Science Advances.]
Glial biology in neurodegenerative diseases reflects a response to, or an induction of, inflammation. Last year, some curious links emerged between inflammation and amyloid. Some scientists reported that interferon-induced transmembrane protein 3, widely expressed in the brain and elevated in aging and AD, binds γ-secretases and ramps up Aβ production (Sep news). Others proposed that phosphorylation of presenilin 1, the catalytic subunit of γ-secretase, drives microglial migration and clearance of amyloid (Aug news).
Vascular System
While scientists have long known that good cardiovascular health equates to good brain health, last year they found that the benefits start in the third decade of life. Obesity in one’s 20s and 30s nearly doubles the risk for late-life dementia, while hypertension and high cholesterol by age 32 increase the likelihood of brain atrophy or cognitive decline, researchers showed at the virtual AAIC (Aug conference news). More evidence now supports the idea that poor cardiovascular health increases risk for vascular dementia, though not amyloidosis (Jan news).
Meanwhile, researchers are making inroads into understanding how blood vessels and the brain’s parenchyma dovetail—a phenomenon called neurovascular coupling. Curiously, this coupling is disrupted by none other than tau. In mice, tau that had migrated from axons displaced nitric oxide synthase in neural dendrites; without dendritic NOS, which produces the vasodilator nitric oxide, blood vessels would not open in response to neural activity (see image below and Aug news).

Muted. The activity-dependent increase in blood flow (color scale) is much greater in control (left) than PS19 tauopathy mice (right). [Courtesy of Park et al., Nature Neuroscience.]
Researchers reported that pericytes, which line capillaries in the brain and form an integral part of the blood-brain barrier, connect to each other via tunneling nanotubes (see image below). Calcium waves traveling through those tubes coordinate dilation of blood vessels near sites of neuronal activation, and constriction of vessels at distant sites. This way, blood flows where it is needed most (Aug news). In larger vessels, endothelial cells and teeny valves do a similar job. Surface indentations on endothelial cells, called calveolae, mediate changes in blood flow in arterioles in response to neural activity, researchers reported, while specialized sphincters, positioned just where arterioles branch, control flow to capillaries (Feb news). Stroking a mouse’s whisker stimulates calveolae and sphincters to dilate blood vessels in the whisker’s corresponding region of the barrel cortex, supplying more blood there.

Mysterious Connection. A tunneling nanotube (above dotted line) surrounded by basement membrane (green) connects the cell body of a proximal pericyte (red) to the process of a distal pericyte on another capillary. [Courtesy of Alarcon-Martinez et al., Nature.]
There is more to endothelial cells and neurodegeneration. According to a large snRNA-Seq study, the AD brain may make more endothelial cells than normal, and their gene expression suggests they are busy building blood vessels and presenting antigens. The finding paints a potentially grim scenario—abnormal capillary growth could open the blood-brain barrier, letting in peripheral immune cells (Oct news). On that score, cytotoxic T cells turned up not just in blood, but also in the brain and CSF of AD patients (Jan news). In the CSF, the T cells multiplied, though no one knows why. Some recognized antigens expressed by Epstein-Barr virus, which is known to infect the CNS.
Ominously, a study suggested that the BBB may be more permeable than has been thought. Receptor-mediated transport ferries large quantities of protein from the blood to the brains of young mice, according to the data, but this slows with age (Jul news). Here again, endothelial cells were important, particularly those lining venules. Implicated in this active transport were the transferrin receptor, the amino acid transporter SLC3A2, the fatty acid transporter MFSD2A, and APOE.
Genetics
Twenty-twenty was not a banner year for AD genetics. While geneticists continue to uncover new risk genes, the emphasis has shifted toward deciphering how they function in disease. Still, the largest GWAS of African Americans to date found one new locus near the insulin-like growth factor receptor gene, and 10 others that fell just below genome-wide significance, including a variant near RBFOX1 (Oct news). A separate study linked this RNA-binding protein gene to AD in a GWAS of amyloidosis (Jun news).
Other loci identified last year included an immunoglobulin G allele that quadrupled risk for AD in people carrying two copies, a variant in angiotensin-converting enzyme that increases risk of AD not by modulating blood pressure but by hastening degeneration of neurons, and a coding variant in the valosin-containing protein gene that causes an autosomal-dominant form of frontotemporal dementia (Aug news; Oct news; Oct news). In an unexpected twist, variants in the Parkinson’s gene LRRK2 were tied to a tauopathy, progressive supranuclear palsy (Mar conference news).
As for functional variants, a few long-known AD genes gave up some secrets. By slowing BACE1 trafficking along axons, GGA3 variants cause axonal swelling that churns out Aβ; PICALM compensates when APOE4 hobbles endocytosis in astrocytes; and the klotho longevity gene appears to preserve memory by holding off tangles in people who have plaques (Nov news; Oct news; Aug news).
Researchers placed phospholipase Cγ2 downstream of TREM2 signaling in microglia. A protective variant in the gene boosts their phagocytosis and lifespan. In a separate pathway triggered by toll-like receptors and intracellular lipid, PLCγ2 stokes inflammation (Jun news; Sep news). For its part, Bin1 was deemed essential for proper release of neurotransmitters and synapse numbers, while SORL1 keeps early endosomes from swelling up, as they do in neurons saddled with familial AD mutations in APP or the presenilins (Mar news; Jun news).
Beyond these single-gene studies, geneticists took a systematic approach to finding functional variants. An epigenetic study used single-nucleotide polymorphisms (SNPs) that are co-inherited with noncoding GWAS variants, and correlated those with active areas of the genome as per a new chromatin map. A focus on disrupted transcription factor binding turned up new functional variants in PICALM and BIN1, as well as RIN3 and STAB1, as candidate AD and PD genes (Oct news).
Other geneticists asked in which cell types AD GWAS hits most likely function—in other words, in which cells do GWAS loci coincide with the most active transcription at that locus? Those would be most affected by risk variants. The study confirmed that microglia express most known AD risk genes, whereas in PD, it was enteric neurons and, surprisingly, oligodendrocytes (May news).
Epigenetic regulators were implicated in neurodegeneration in 2020. Mutations in the DNA demethylase 10-11 translocation 2 (TET2) double a person’s risk for AD and FTD (May news). Carriers were predicted to decline much faster on the clinical dementia rating scale (see image below). In related epigenetic news, researchers found that levels of BAZ-2, which recruits transcription factors to modified histones, and SET-6, a histone3/lysine9 (H3K9) methyltransferase, increase in the brain as animals age. The two suppress mitochondrial gene expression and accelerate memory loss in mice. In people, levels of the two epigenetic modulators correlated with AD progression (Feb news).
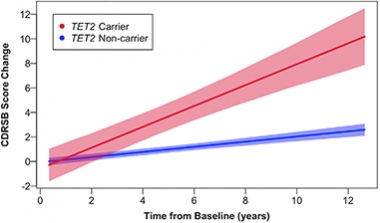
Worsening Fast. Extrapolations predict that TET2 carriers will decline faster than noncarriers, both cognitively and functionally. [Courtesy of Cochran et al., American Journal of Human Genetics.]
Might all the common risk variants together cause even more trouble? In October, researchers reported that people who had the highest polygenic risk scores had a smaller hippocampus—even as young adults. ApoE accounted for most of the risk (Oct news). Polygenic scores even suggest that genetic risk for AD in women is different than in men. When scientists calculated hazard scores based on sex-specific GWAS data, they better predicted outcomes. In other words, female polygenic scores better predicted AD in women than in male scores, and vice versa (Jul news). This, plus news about the X chromosome, is finally beginning to make sense of perplexing epidemiology data showing that while women are more likely to get AD than are men, they also survive longer with it.
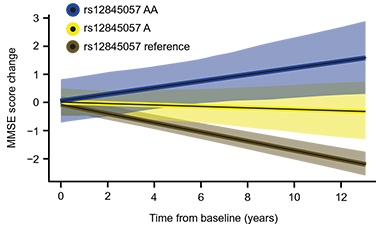
Resilience Variant. Carriers of two (blue) or one (yellow) copy of a variant that boosts KDM6A expression decline more slowly on the mini mental state exam than noncarriers (brown). [Courtesy of Davis et al., Science Translational Medicine, 2020.]
An explanation for the latter came in August, when researchers reported that among four sex genotypes, i.e. X, XX, XY, and XXY, having two X chromosomes protected mice from amyloidosis and memory loss, irrespective of the Y chromosome. This seems to settle the question of whether women’s extra X or lack of Y explains their apparent resilience in dementia. But how could a second X help when it is normally silenced? X inactivation ensures the additional X does not double the dose of X transcripts. As it turns out, some genes escape this quality-control step and are transcribed. One, called KDM6A, encodes a histone demethylase that profoundly affects gene transcription. It is elevated in AD, possibly as a compensatory response. When expressed in men, it protects them from amyloid and memory loss, too (see image above and Aug news).
Air Pollution
Tenuous links between air pollution and neurodegeneration have been wafting up from the literature for decades, but recently scientists started studying this epidemiology more systematically. New findings have linked particulate matter to neurodegeneration and cognitive decline, and not just via effects on the cardio-respiratory system. Evidence suggests that particulate matter can get into the brain, possibly through olfactory nerves (May news; May news).
In December, leaders of the IDEAS study, which was designed to test whether amyloid PET scans improve disease management, published a pollution sub-study. They found that people who live in locations with the highest levels of PM2.5, i.e., particulates smaller than 2.5 microns in diameter, had more amyloid plaques and more neurodegeneration than those who breathed cleaner air (see image below and Dec news).

Particular About Air. Estimated air pollution where IDEAS study participants lived from 2002–2003 (left) and 2015–2016 (right) as mean daily PM2.5 levels. [Courtesy of Iaccarino et al., JAMA Neurology.]
The evidence has grown such that this year, a Lancet Commission, in updating its “Dementia prevention, intervention, and care” report of 2017, added air pollution as a modifiable risk factor for AD, calculating that it accounts for 2 percent of dementia cases (Aug conference news).
In a unique case of pollution, it appears that people who cleaned up the World Trade Center site in Manhattan are showing signs of dementia in their 50s. Their cognitive impairment correlated with exposure to toxic dust and with post-traumatic stress (Jan news). Exactly what this means remains murky, but scientists plan to follow these responders to see if their conditions are progressive or temporary.
Is Alzheimer’s a Contagion?
No, not of SARS-CoV-2, but of proteopathic seeds of amyloidogenic proteins. Found in pituitary extracts used to prepare human growth hormone, and in brain dura mater from cadavers, toxic protofibrils of Aβ can bring on a type of cerebral amyloid angiopathy decades later in young-adult growth hormone or dural graft recipients. While these protocols are no longer used, concerns were raised that seeds of Aβ, α-synuclein, or tau might be travel from brain to brain on neurosurgery tools.
In the fall, two advisory groups weighed in. In a white paper, 37 leading experts in the EU concluded no evidence existed for such transmission during neurosurgery. All the same, they urged surgeons to use separate instruments for adults and children, and called for a long-term epidemiological study (Sep news). A panel convened by the National Institute on Aging in the U.S. agreed, noting that research was inadequate and calling for better characterization of proteopathic seed infectivity, stability, and distribution in the body (Oct news).
As for current global contagion, it appears that just as transmission and deaths are at their highest, vaccines are riding to the rescue. There’s now hope that by mid-year, the worst will be behind us. On this note, let’s all raise a toast to science.—Tom Fagan and Gabrielle Strobel
References
News Citations
- Coronavirus Takes Its Toll on Alzheimer’s Clinical Studies
- A ‘Perfect Storm’ for Families Grappling with Dementia
- Social Distancing Shutters Alzheimer’s Research
- Virtual Meetings Try Out Different Formats
- Alzheimer’s Research Restarts, Cautiously
- In the Wake of COVID-19: Trials Interrupted, Delayed, Cancelled
- Biogen Asks FDA To Approve Aducanumab
- FDA Advisory Committee Throws Cold Water on Aducanumab Filing
- Aducanumab Still Needs to Prove Itself, Researchers Say
- BAN2401 Forges AHEAD into Phase 3, Preclinical AD
- TRC-PAD Funnel Finally Touches Down
- In DIAN-TU, Gantenerumab Brings Down Tau. By a Lot. Open Extension Planned
- New Data from Past BACE Inhibitor Trials Shed Light on Side Effects
- Low-Dose BACE Inhibitors Might Preserve Synaptic Function
- Mounting Modifications Move Tau Toward Aggregation in Alzheimer’s Brain
- Active Tau Vaccine: Hints of Slowing Neurodegeneration
- α-Synuclein Antibody Misses Primary, May Have Signal on Secondaries
- Blunt Instruments: α-Synuclein Antibodies Poorly Distinguish Forms
- Plasma Aβ Test Wins Approval—Are p-Tau Tests Far Behind?
- A Phospho-Tau Plasma Assay for Alzheimer’s?
- Plasma p-Tau217 Set to Transform Alzheimer’s Diagnostics
- In Colombian Alzheimer’s Kindred, Blood NfL Climbs 22 Years Before Symptoms
- Plasma NT1: This Tau Snippet Predicts Cognitive Decline in Alzheimer’s
- MTBR-243 Tau: A Fluid Biomarker for Tangles Themselves?
- Different CSF Phospho-Taus Match Distinct Changes in Brain Pathology
- Peter Davies, Beloved Giant of Alzheimer’s Disease Research, Dies at 72
- CryoEM of CBD Tau Suggests Another Unique Protofibril
- Behold the First Human α-Synuclein CryoEM Fibril Structure
- Paper Alert: CryoEM Structures of α-Synuclein Published
- Molecular Structure of LRRK2 Gives Clues to Parkinson’s
- Better than Crystal Clear? Cryo-EM Achieves Atomic Resolution
- Tau PET: The Field Expands Rapidly
- Primary Tauopathies Get New PET Ligands
- PET Tracer PI-2620 Detects 4R Tau Deposits
- Could Tau PET Replace Amyloid Biomarkers as a Diagnostic for AD?
- New at Tau2020: PET Detects First Traces of Tangles in Rhinal Cortex
- Connectivity, Not Proximity, Predicts Tau Spread
- Simulating Tangle Spread Along Axons, Scientists Predict Tau PET Patterns
- Tangle Density Foretells How Fast a Person’s Brain Will Shrink
- Individualized Tau PET Model Outperforms Predictive Power of Braak Staging
- How Much Amyloid Will Kick Off Tangles, and Decline?
- IDEAS Finds Small Drop in Hospitalizations, Missing Goal
- PET Tracer Detects Synapse Loss Across Alzheimer’s Brain
- Multimodal Imaging of Neurodegenerative Diseases Links Pathology and Cellular Dysfunction
- When It Comes to Alzheimer’s Disease, Do Human Microglia Even Give a DAM?
- Human and Mouse Microglia React Differently to Amyloid
- Single-nucleus RNA Sequencing Misses Activation of Human Microglia
- Most Detailed Look Yet at Activation States of Human Microglia
- Paper Alert: Those PIGs! Spatial Transcriptomics Add Human Data
- Clocking In: Timestamped RNA Charts Transcriptional History
- Massive Proteomics Studies Peg Glial Metabolism, Myelination, to AD
- Neuronal SRPX2 Spoils Microglial Appetite for Synapses
- With IL-33, Neurons Tempt Microglia to Nibble At Synapses
- Too Phatal: How Microglia, Astrocytes Snuff Out Dying Neurons
- IFITM3 Forges Link Between Neuroinflammation and Aβ Production
- Does Phosphorylation of Presenilin 1 Drive Microglial Migration, Aβ Clearance?
- Heart Health Is Brain Health, and It Starts in Your 20s
- Vascular Dysfunction Taxes Cognition, but Not Via Amyloid, AD
- With Tau in Synapses, NO Neurovascular Coupling
- Tunneling Nanotubes—How Pericytes Control Blood Flow
- Ruffles and Sphincters Control the Spigot of Fresh Blood in the Brain
- Do Endothelial Cells Spur Capillaries to Grow in Alzheimer’s Brain?
- Attack of the Clones? Memory CD8+ T Cells Stalk the AD, PD Brain
- Blood-Brain Barrier Surprise: Proteins Flood into Young Brain
- Largest Alzheimer GWAS in African Americans Finds New Variants
- RBFOX1 Gene Tied to Amyloidosis in Preclinical Alzheimer’s
- Can an Antibody Allele Boost Your Alzheimer’s Risk By Giving Herpes an Edge?
- New ACE Variant Speeds Neurodegeneration in Alzheimer’s Mice
- VCP Coding Mutation Causes a Tauopathy With Vacuoles
- Tau2020: Meeting for Tauopathies Debuts Genetic Variants
- Paper Alert: Trafficking Variants Cause BACE Buildup in Axons
- In Astrocytes, ApoE4 Bungles Endocytosis, PICALM Picks Up the Slack
- Klotho VS Variant Preserves Memory by Preventing Tangles
- Janus-Faced PLCγ2? Alzheimer’s Risk Protein Toggles TREM2 and TLR Pathways
- Protective AD Variant Pinpoints Sweet Spot for Microglial Activation
- Alzheimer’s Gene BIN1 Promotes Synaptic Transmission
- Without SORL1, Endosomes Swell in Neurons but not Microglia
- Epigenomic Roadmap Points to Causal Genes
- Which Cell Types Execute Your Genetic Risk? For AD and PD, Scientists Know.
- Mutations in Epigenetic Gene Linked to Neurodegeneration
- In Aging, Epigenetic Wet Blanket Douses Mitochondria
- Alzheimer's Risk Genes Nip at Hippocampus Throughout Life
- Women and Men Differ in Their Genetic Risk for Alzheimer’s Progression
- Does Second X Chromosome Boost Women’s Resilience Against Alzheimer’s?
- Air Pollution and Dementia—Through Hazy Data, Links Emerge
- The Air We Breathe—How Might Pollution Hurt the Brain?
- You Are What You Breathe: Polluted Air Tied to Plaques, Brain Atrophy
- Lancet Commission’s Dementia Hit List Adds Alcohol, Pollution, TBI
- Do World Trade Center Responders Get Early Onset Plaques and Tangles?
- Could Contaminated Scalpels Seed Amyloidosis?
- NIA Weighs in on Neurodegenerative Disease Transmissibility
Therapeutics Citations
- Nuplazid
- Leqembi
- Gantenerumab
- Donanemab
- UB-311
- ALZ-801
- GV-971
- Azeliragon
- Epigallocatechin Gallate (EGCG)
- CT1812
- Varoglutamstat
- Semorinemab
- Tilavonemab
- Gosuranemab
- Zagotenemab
- JNJ-63733657
- Bepranemab
- BIIB076
- E2814
- Lu AF87908
- PNT001
- AADvac1
- ACI-35
- AL002
- Inzomelid
- Pepinemab
- Neflamapimod
- DNL343
- Vafidemstat
- Prasinezumab
- DNL201
- LTI-291
- NPT520-34
Other Citations
External Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.