Barriers Between Blood and CSF, Brain Yield to Aβ—Not a Bad Thing?
Quick Links
The barrier between the blood and central nervous system crumbles in Alzheimer’s disease, but researchers have known little about how this happens, or what it does to brain pathology. Two new papers shed some light on how Aβ damages the cells that protect the brain parenchyma and cerebrospinal fluid. The studies examine different systems and describe distinct mechanisms, but both add to the picture of what may happen in disease.
One study appeared in the September 18 Science Advances, an open-access online journal AAAS launched this past February. Researchers led by Matthew Campbell at Trinity College Dublin report that Aβ40 monomers open up small gaps in the blood-brain barrier, allowing the peptides to squeeze between endothelial cells out into the bloodstream. This clearance route was not previously suspected, and may help the brain eliminate high concentrations of Aβ, Campbell suggested. In keeping with this, he found that transiently weakening the integrity of the blood-brain barrier in mouse models of amyloid accumulation heightened Aβ clearance, lowered brain amyloid load, and improved cognition, hinting that this mechanism could be harnessed therapeutically. Campbell and colleagues are testing this idea in primates.

Layered Barrier.
Tight junctions between capillary endothelial cells form the blood-brain barrier, with pericytes (blue) and astrocytes (peach) providing additional layers of protection not found in the blood CSF barrier. [Courtesy of Science Advances/AAAS.]
By contrast, researchers led by Roosmarijn Vandenbroucke at Ghent University, Belgium, focused on harmful effects of Aβ on the choroid plexus epithelium, a membrane that lines brain ventricles and separates blood vessels from the cerebrospinal fluid (CSF). In the September 16 Journal of Neuroscience, these researchers reported that high levels of synthetic Aβ42 oligomers damaged this barrier within hours, disrupting tight junctions between epithelial cells and distorting the cells’ shape. The scientists traced the cause to upregulated inflammatory factors and matrix metalloproteinases. MMPs are enzymes that chew up proteins, including those in tight junctions. Inhibition of MMPs prevented damage.
Costantino Iadecola at Weill Cornell Medical College, New York, commended the authors for focusing on an area that receives little study. “We don’t know enough about the blood-brain barrier and blood-CSF barrier. These papers address interesting questions that should improve our understanding of Aβ’s effects on these structures,” he told Alzforum.
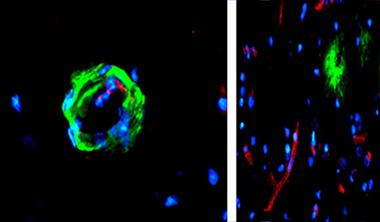
Vessel Plaques Loosen Junctions?
Amyloid-laden (green) capillary (left, cross-section) expresses sparse tight junction protein occludin (red). Occludin is abundant in plaque-free vessels nearby (right, lower magnification). [Courtesy of Science Advances/AAAS.]
The blood-brain barrier forms in capillaries, where proteins such as claudin-5 and occludin bind to create tight junctions between the endothelial cells that comprise the vessel. The junctions prevent molecules from slipping between the cells, thus blocking passage of solutes into or out of the brain. This barrier helps maintain brain homeostasis but also complicates clearance of detritus, including Aβ. Some Aβ can pass through the blood-brain barrier via active transport across endothelial cells (see Deane et al., 2003; Deane et al., 2004). On the other hand, it is still unclear whether Aβ can also filter out through the tight junctions between endothelial cells, the so-called “paracellular” pathway. Intriguingly, Aβ has been found to suppress tight junction proteins (see Sep 2011 news; Carrano et al., 2011; Hartz et al., 2012).
Confirming these latter findings, first author James Keaney in Campbell’s group demonstrated that adding synthetic Aβ40 monomers and dimers transiently suppressed claudin-5 and occludin in endothelial cell cultures. Similarly, in aged Tg2576 mice, which overexpress amyloid precursor protein, the authors saw scant claudin-5 and occludin along portions of capillaries clogged with amyloid. Postmortem brain sections from 30 Alzheimer’s patients and controls also showed less claudin-5 and occludin in blood vessels choked with amyloid, but normal amounts in healthy vessel sections just a few microns away (see image below).
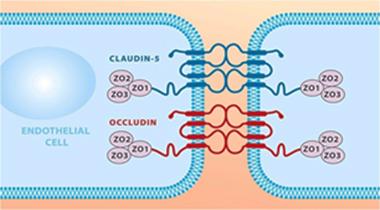
Mind the Gap.
The tight-junction proteins claudin-5 and occludin prevent transport between endothelial cells. [Courtesy of Science Advances/AAAS.]
Could the drop in these tight-junction proteins allow Aβ40 to slip out of the brain? The authors used siRNAs to suppress claudin-5 and occludin translation in endothelial cell cultures that maintain tight junctions and form a monolayer separating two pools of medium. The authors found that in this cell-based model of the blood-brain barrier, Aβ40 monomers, but not dimers, passed through the weakened barrier. This suggested the gaps were only large enough for molecules less than 8 kDa in size to pass. Suppression of the same tight-junction proteins in wild-type mice allowed molecules of 3 kDa, but not 10 kDa, to diffuse from brain to blood.
The authors wondered what effect this permeability would have in the context of excess amyloid. They suppressed claudin-5 and occludin expression once every three weeks for nine months in Tg2576 and APPPS1 mice, causing a sporadic, transient relaxation of the blood-brain barrier. Plasma levels of soluble Aβ40 jumped about threefold, while brain levels dropped in half. Treated animals performed better in a T-maze, which measures spatial memory.
The results hint at therapeutic potential, suggested the author. Campbell is investigating whether suppressing these tight-junction proteins in mice just before administering an anti-amyloid antibody would enhance Aβ clearance from brain. Campbell noted that so far, the transient suppression of tight-junction proteins by siRNA appears safe in both mice and monkeys, with no signs of the brain edema that is known to accompany blood-brain barrier breakdown. He believes this is because the barrier opens only slightly, for a brief period. “We are taking away just a small amount of junctional material to render the barrier marginally permeable,” Campbell explained.
Others agreed the finding has promise. “The data indicate you can modulate the absorption of Aβ into blood, and that in turn lowers brain amyloid and delays cognitive decline. That’s worth investigating,” noted Roy Weller at the University of Southampton, U.K. However, researchers cautioned that side effects would have to be carefully studied to ensure toxic molecules are not entering the brain through the weakened barrier.
Vandenbroucke called the finding fascinating, though she wondered how systemic suppression of tight-junction proteins would affect the blood-CSF barrier. Molecules that pass through that different boundary gain direct access to CSF and brain interstitial fluid, unlike molecules that wriggle through the blood-brain barrier, where other cell types such as pericytes and astrocytes provide an additional layer of protection for brain parenchyma. Thus, the blood-CSF barrier might be more vulnerable than the blood-brain barrier to harmful effects from transient permeability, Vandenbroucke speculated.
The barrier between blood and CSF has received far less attention than the blood-brain barrier, though some studies claim that this boundary also falters in AD brain (see Serot et al., 2000; Krzyzanowska and Carro, 2012). To investigate, joint first authors Marjana Brkic and Sriram Balusu in Vandenbroucke’s group injected synthetic Aβ42 oligomers into the brain ventricles of wild-type mice. After both two hours and six hours, they saw a massive boost in the expression of inflammatory cytokines IL6, IL1β, and TNFα, in the choroid plexus. This was accompanied by increased expression and activity of several MMPs, in particular MMP3, which shot up 15-fold. At the same time, epithelial cells in the choroid plexus shrank, losing their plump, cuboidal shape. Expression of claudin-5 and occludin dropped in half, and the blood-CSF barrier leaked.
MMPs have been implicated in blood-CSF barrier disruption before (see Zeni et al., 2007; Batra et al., 2010; Vandenbroucke et al., 2012). Brkic and colleagues wondered if these enzymes, which are triggered by inflammation, might be chewing up the barrier. Supporting this, co-injecting a broad-spectrum MMP inhibitor along with Aβ42 prevented barrier damage. Likewise, MMP3 knockout mice injected with Aβ42 oligomers developed only slight leakage compared to injected wild-type counterparts.
These experiments highlight acute effects of Aβ, but what happens long-term? In ongoing work, the authors are examining the blood-CSF barrier days and weeks after Aβ injection. They are also studying this barrier in APPPS1 mice, which overproduce Aβ. In preliminary work, these animals appear to spontaneously develop some leakage between CSF and blood, though less pronounced than in injected mice, Vandenbroucke told Alzforum. This might mean that under chronic conditions of high Aβ, tight junctions can stabilize to some extent, she suggested.
In addition, Vandenbroucke will assess whether inhibiting MMPs improves cognition in mice. Such data would make a case for therapeutically suppressing these enzymes. Broad-spectrum MMP inhibitors have been tested in clinical trials for cancer, but caused side effects such as muscle pain, and are not an option, Vandenbroucke noted. Alternatives would be to target MMP3 specifically, or to suppress upstream inflammatory factors that boost its expression. Alzheimer patients’ brains have been reported to contain elevated levels of metalloproteinases (see Wang et al., 2014).
Berislav Zlokovic at the University of Southern California, Los Angeles, found the data intriguing. "The present study is novel in shedding light on the negative impact of Aβ1-42 oligomers at the BCSFB and the potential ability of MMPs, specifically MMP-3, to mediate this effect. MMPs appear to be crucially important in regulating the integrity of the brain’s specialized vascular barriers, including both the blood-brain barrier and blood-CSF barrier,” he wrote to Alzforum (see full comment below).—Madolyn Bowman Rogers
References
News Citations
Research Models Citations
Paper Citations
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, Kindy M, Stern D, Zlokovic B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003 Jul;9(7):907-13. PubMed.
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004 Aug 5;43(3):333-44. PubMed.
- Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011 Sep 1;15(5):1167-78. PubMed.
- Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, Mihaljevic I, Bogdahn U, Klünemann HH, Schuierer G, Schlachetzki F. Amyloid-β Contributes to Blood-Brain Barrier Leakage in Transgenic Human Amyloid Precursor Protein Mice and in Humans With Cerebral Amyloid Angiopathy. Stroke. 2012 Feb;43(2):514-23. PubMed.
- Serot JM, Béné MC, Foliguet B, Faure GC. Morphological alterations of the choroid plexus in late-onset Alzheimer's disease. Acta Neuropathol. 2000 Feb;99(2):105-8. PubMed.
- Krzyzanowska A, Carro E. Pathological alteration in the choroid plexus of Alzheimer's disease: implication for new therapy approaches. Front Pharmacol. 2012;3:75. Epub 2012 May 3 PubMed.
- Zeni P, Doepker E, Schulze-Topphoff U, Schulze Topphoff U, Huewel S, Tenenbaum T, Galla HJ. MMPs contribute to TNF-alpha-induced alteration of the blood-cerebrospinal fluid barrier in vitro. Am J Physiol Cell Physiol. 2007 Sep;293(3):C855-64. Epub 2007 May 16 PubMed.
- Batra A, Latour LL, Ruetzler CA, Hallenbeck JM, Spatz M, Warach S, Henning EC. Increased plasma and tissue MMP levels are associated with BCSFB and BBB disruption evident on post-contrast FLAIR after experimental stroke. J Cereb Blood Flow Metab. 2010 Jun;30(6):1188-99. Epub 2010 Mar 3 PubMed.
- Vandenbroucke RE, Dejonckheere E, Van Lint P, Demeestere D, Van Wonterghem E, Vanlaere I, Puimège L, Van Hauwermeiren F, De Rycke R, Mc Guire C, Campestre C, López-Otin C, Matthys P, Leclercq G, Libert C. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci. 2012 Jul 18;32(29):9805-16. PubMed.
- Wang XX, Tan MS, Yu JT, Tan L. Matrix metalloproteinases and their multiple roles in Alzheimer's disease. Biomed Res Int. 2014;2014:908636. Epub 2014 Jun 24 PubMed.
Further Reading
News
- ApoE4 Makes Blood Vessels Leak, Could Kick Off Brain Damage
- Stop, Hey, What’s That Sound? ... Amyloid Is Going Down?
- Does Aggregated Aβ Pass Directly From Blood to Brain?
- Brain Drain—“Glymphatic” Pathway Clears Aβ, Requires Water Channel
- Glymphatic Flow, Sleep, microRNA Are Frontiers in Alzheimer’s Research
- Fluid Markers and Imaging Back Idea of Breached Blood-Brain Barrier
- In Aging Brain, Blood-Brain Barrier Starts Leaking in Hippocampus
Primary Papers
- Keaney J, Walsh DM, O'Malley T, Hudson N, Crosbie DE, Loftus T, Sheehan F, McDaid J, Humphries MM, Callanan JJ, Brett FM, Farrell MA, Humphries P, Campbell M. Autoregulated paracellular clearance of amyloid-β across the blood-brain barrier. Sci Adv. 2015 Sep 4;1(8):e1500472. PubMed.
- Brkic M, Balusu S, Van Wonterghem E, Gorlé N, Benilova I, Kremer A, Van Hove I, Moons L, De Strooper B, Kanazir S, Libert C, Vandenbroucke RE. Amyloid β Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J Neurosci. 2015 Sep 16;35(37):12766-78. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
University of Southern California
University of Southern California, Keck School of Medicine
University of Southern California
The brain’s microenvironment is strictly maintained by specialized highly vascularized barriers, including the blood-brain barrier (BBB) in the brain parenchyma and the blood-cerebrospinal fluid barrier (BCSFB) in the choroid plexus. Brkic et al. present an interesting study to evaluate the impact of synthetic human Aβ1-42 oligomers on BCSFB integrity in mice. The BCSFB, unique from the BBB due to its epithelial monolayer and fenestrated endothelium, contributes to central nervous system homeostasis by producing cerebrospinal fluid (CSF) and restricting the passage of undesirable molecules and pathogens into the brain, as well as by clearing metabolic waste products from CSF.
This study found that Aβ1-42 injected mice have increased metalloproteinase (MMP) enzymatic activity and increased mRNA levels of MMP-3, in addition to morphological changes of choroid plexus epithelial cells and increased BCSFB permeability. These findings suggest that Aβ1-42 oligomers detrimentally impact BCSFB integrity, which can cause subsequent disruption of homeostasis of the neuronal internal milieu. Such disruption may also alter CSF production and impair Aβ clearance from the brain via the interstitial fluid (ISF)-CSF route.
This study further reports that disrupted BCSFB permeability in response to Aβ1-42 oligomers can be reversed by the administration of a general MMP inhibitor as well as in Mmp3-/- mice. Overall, the present study is novel in shedding light on the negative impact of Aβ1-42 oligomers at the BCSFB and the potential ability of MMPs, specifically MMP-3, to mediate this effect. MMPs appear to be crucially important in regulating the integrity of the brain’s specialized vascular barriers, including both blood-brain barrier and blood-CSF barrier. At the BBB, for example, ApoE4 induces upregulation of MMP-9 in brain capillary pericytes that promotes BBB breakdown (Bell et al., 2012). The extent to which pericyte injuries can contribute to changes in BCSFB permeability remains, however, elusive at present, as well as the source of elevated MMP-3 activity, which all could present interesting directions for future studies.
References:
Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012 May 24;485(7399):512-6. PubMed. Correction.
University of South Alabama
University of Southern California
Impaired clearance of Aβ is a key contributing factor to Aβ accumulation in the brain and the progression of AD. Studies focusing on Aβ clearance mechanisms, regulation of Aβ efflux, and ways to modulate Aβ removal are of importance to the field. The blood-brain barrier, comprised of a tight monolayer of cerebral endothelial cells, authorizes molecules to enter and leave the brain. Keaney et al. present an interesting study using a systemic siRNA approach to manipulate expression of tight-junction proteins at the site of the blood-brain barrier, and determine how this affects the blood-brain barrier molecular permeability.
The authors hypothesize that a controlled opening of the blood-brain barrier by downregulation of tight junction proteins—claudin-5 and occludin—permits a paracellular clearance of Aβ across the blood-brain barrier. They suggest a method for size-selective removal of Aβ facilitated by RNAi-mediated co-suppression of the two transmembrane tight-junction proteins. Opening the blood-brain barrier “on demand” is a difficult task, and if achieved successfully might aid in both the delivery of neuropharmaceuticals to the brain that otherwise cannot cross the blood-brain barrier and/or enhance clearance of potentially toxic products from the brain that contribute to brain pathology. However, the concept of “autoregulation”—a phenomenon where the brain itself enables paracellular clearance of Aβ during AD pathology—will need to be examined carefully and in relation to other multiple Aβ clearance mechanisms that exist physiologically. These mechanisms include the transvascular clearance across the blood-brain barrier, interstitial fluid bulk flow, i.e., traditional perivascular clearance, glymphatic paravascular clearance, cerebrospinal fluid absorption, and enzymatic degradation (Ramanathan et al., 2015).
The authors suggest that they can open the blood-brain barrier in a size-specific manner and, for example, allow only small molecules that are less than 10 kDa to pass. Future studies should proceed with caution because opening the blood-brain barrier may facilitate the entry of toxic blood-derived products, pathogens, and blood cells into the brain that have been shown to contribute to pathogenesis and neurodegenerative changes in monogenic rare human diseases, and may also influence the disease process in complex neurological diseases such as Alzheimer’s disease and amyotrophic lateral sclerosis.
References:
Ramanathan A, Nelson AR, Sagare AP, Zlokovic BV. Impaired vascular-mediated clearance of brain amyloid beta in Alzheimer's disease: the role, regulation and restoration of LRP1. Front Aging Neurosci. 2015;7:136. Epub 2015 Jul 15 PubMed.
UBC
Both these very interesting studies (Keaney et al., 2015, and Brkic et al., 2015) build on our discovery that the blood-brain barrier is perturbed concomitantly with the initiation of new cerebral vessels in AD. During angiogenesis, which entails endothelial cell division, the tight junctions that mediate the barrier between the endothelial cells in the BBB are programmed to reorganize, barrier function is reduced, and permeability increased. It appears that amyloid(s) is the trigger for the angiogenesis we observe in both mice and humans with AD.
Similar to Keaney et al., our earlier studies interpreted the BBB’s age-dependent breakdown and its increased permeability as being an impairment that allows the diffusion of proteins through the barrier (Ujiie et al., 2003; Dickstein et al., 2006).
However, that breakdown and permeability increase now appears to be a result of endothelial cell division rather than cellular deterioration (Biron et al., 2011; Biron et al., 2013).
We also observed BBB angiogenesis in postmortem tissues from several pathology-confirmed cases of AD. Thus, these studies likely explain the mechanism that underpins the observations in Keaney et al.
We and others interpret this brain endothelial cell division as being driven by amyloid(s). Elegant studies in zebrafish confirm this (Cameron et al., 2012).
Comparing the two recently published studies, one group studied brain endothelium (Keaney et al., 2015), the other epithelial cells in the choroid plexus (Brkic et al., 2015). These are different cell types, though it is perhaps interesting to ponder that amyloids appear to be triggering cell division rather than deterioration of either barrier.
References:
Ujiie M, Dickstein DL, Carlow DA, Jefferies WA. Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation. 2003 Dec;10(6):463-70. PubMed.
Dickstein DL, Biron KE, Ujiie M, Pfeifer CG, Jeffries AR, Jefferies WA. Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J. 2006 Mar;20(3):426-33. PubMed.
Biron KE, Dickstein DL, Gopaul R, Jefferies WA. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer's disease. PLoS One. 2011;6(8):e23789. PubMed.
Biron KE, Dickstein DL, Gopaul R, Fenninger F, Jefferies WA. Cessation of neoangiogenesis in Alzheimer's disease follows amyloid-beta immunization. Sci Rep. 2013;3:1354. PubMed.
Cameron DJ, Galvin C, Alkam T, Sidhu H, Ellison J, Luna S, Ethell DW. Alzheimer's-related peptide amyloid-β plays a conserved role in angiogenesis. PLoS One. 2012;7(7):e39598. PubMed.
Make a Comment
To make a comment you must login or register.