Astrocytes, In Addition to Neural Networks, May Stoke Synuclein Pathology
Quick Links
When α-synuclein fibrils spark pathology in the brain, the path of destruction may stray from well-trodden neural superhighways, according to a paper in the May 29 Molecular Neurodegeneration. Researchers led by Paramita Chakrabarty and Benoit Giasson at the University of Florida in Gainesville reported that following injection of α-synuclein fibrils into transgenic mice overexpressing the human protein, pathology sprouted throughout the brain, even into regions with no apparent anatomic connections to the injection site. A closer look at the synuclein pathology revealed that much of it resided inside astrocytes. The researchers concluded that these glial cells may play an underappreciated role in spreading α-synuclein pathology.
Commentators and the researchers themselves pointed out caveats with the study, including that astrocytes in the transgenic animals highly expressed α-synuclein, normally a predominantly neuronal protein. Still, Giasson and Chakrabarty said the findings highlight a potential role of glia in the propagation of synuclein pathology.
Researchers believe α-synuclein and other misfolded amyloidogenic proteins use a prion-like templating mechanism to creep across the brain, corrupting normally folded protein as they go. Studies led by Virginia Lee at the University of Pennsylvania reported that preformed, sonicated α-synuclein fibrils injected into the striata or neocortices of mice expressing human mutant α-synuclein seeded pathology in neuroanatomically connected regions, and that this occurs in wild-type mice as well (see Apr 2012 news; Nov 2012 news). Since then, multiple studies have demonstrated the same phenomenon in wild-type and transgenic mice, rats, even marmosets (see Masuda-Suzukake et al., 2014; Paumier et al., 2015; Jun 2015 news; Dec 2015 conference news; Breid et al., 2016; Shimozawa et al., 2017).
However, Giasson, working with researchers in Todd Golde’s lab, also at the University of Florida, reported that α-synuclein fibrils—prepared using a slightly different method than Lee’s—triggered proteopathic spread only in transgenic mice overexpressing human mutant α-synuclein; they failed to move beyond the injection site, in this case the hippocampus or cortex, in non-transgenic mice (see Apr 2014 news).
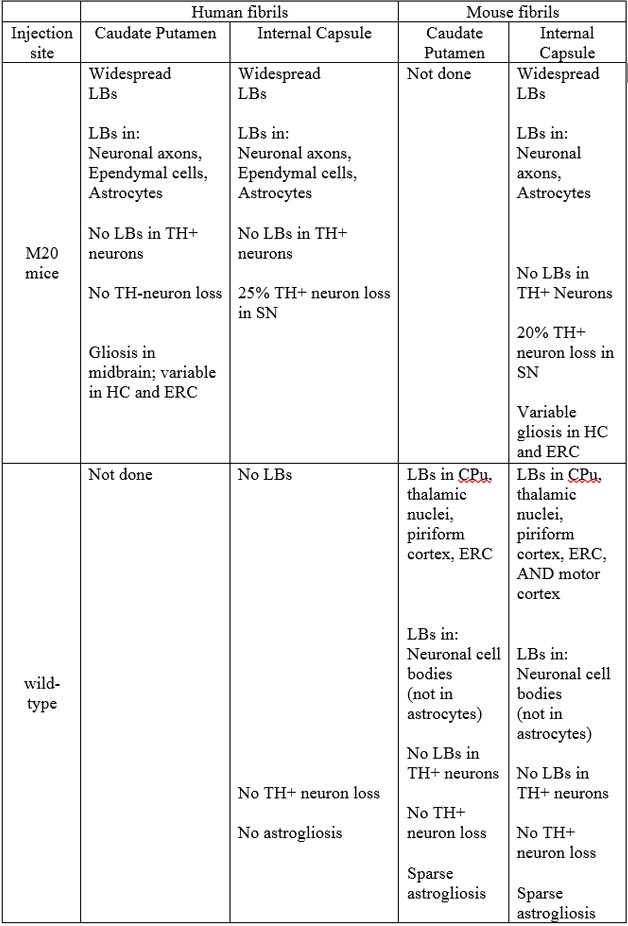
Tracking Pathology. Table lists the sites of synuclein and other pathologies evoked by injected synuclein fibrils. (SN, substantia nigra; HC, hippocampus; ERC, entorhinal cortex.)
In the current study, these researchers teamed up with Chakrabarty’s lab to further investigate the nature of the proteopathic spread and assess the role of glial cells in the process. First author Zachary Sorrentino, a graduate student in Giasson’s lab, used M20 mice; they express human wild-type α-synuclein under control of the mouse prion promoter, which is active in neurons and in glial cells. Giasson helped create these animals as a postdoc in Lee’s and John Trojanowski’s labs. They overexpress the protein about fivefold over endogenous levels. However, unlike mice that express the A53T mutant version of α-synuclein controlled by this promoter, M20 develop neither synuclein pathology nor neurodegeneration (see May 2002 news). Sorrentino injected fibrils of either human or mouse α-synuclein into the caudate putamen (CPu) of eight-week-old M20 mice. This gray-matter region in the dorsal striatum makes extensive contacts to the dopaminergic neurons in the substantia nigra, which fall prey to Parkinson’s disease (PD). In a separate group of M20 mice, he injected fibrils into the internal capsule (IC), a white-matter tract that intersects the CPu and serves as a conduit between motor areas, the brain stem, and cerebellar regions, and also between the thalamus and prefrontal cortex.
Four to five months later, Sorrentino and colleagues euthanized the mice and analyzed sections of their brains for α-synuclein inclusion pathology. They used α-synuclein antibodies specific for phosphorylated and/or misfolded α-synuclein, as well as antibodies specific for p62, a marker of Lewy body (LB)-like inclusions. Whether the animals had been injected in the CPu or IC, LB-like α-synuclein inclusions spread throughout the brain. While some pathology-laden regions clearly had contacts to the CPu and IC, others had no known neuroanatomical connection to these areas. Animals injected with human α-synuclein had slightly more extensive pathology than those injected with mouse α-synuclein fibrils. Pathology appeared in axons and neuronal cell bodies. In addition, at least in animals injected with human fibrils, it also collected in ependymal cells—glial cells that line the CSF-filled lateral ventricles (see table above).
A different picture emerged when the researchers injected wild-type mice with α-synuclein fibrils. In animals injected with human fibrils, no LB-like inclusions emerged. In those injected with mouse fibrils, pathology arose in some, but not all, regions directly connected to the injection site. Inclusions cropped up in the dorsal striatum, thalamic nuclei, piriform cortex, and entorhinal cortex in animals injected in the CPu or IC, but not in the substantia nigra or hypothalamus. Additionally, IC-injected mice also accumulated pathology in the motor cortex, likely owing to the white-matter tract’s extensive connections to motor areas. In these non-transgenic mice, pathology appeared restricted to neuronal cell bodies, with little observed in axons and none in ependymal cells. This contrasts with Giasson’s own 2014 findings, in which α-synuclein inclusions only appeared in the vicinity of the injection site. In that paper, animals received injections into the hippocampus or cortex, rather than the CPu or IC. Giasson said that he does not know what’s behind this difference in spread.
The researchers next investigated whether the induced α-synuclein inclusions had taken a neurodegenerative or inflammatory toll on the animals. In non-transgenic mice, the number of tyrosine hydroxylase (Th)-positive dopaminergic neurons in the substantia nigra was normal, regardless of where mouse α-synuclein fibrils were injected. These mice had sparse astrogliosis, confined to areas with synuclein pathology. In M20 mice injected in the IC with human or mouse fibrils, the number of dopaminergic neurons nudged down by 25 and 20 percent, respectively. Gliosis primarily flared up in areas distal from the injection site—in the hippocampus and entorhinal cortex. Curiously, M20 mice injected in the CPu lost no neurons, despite having robust gliosis throughout the midbrain. The researchers did not perform control injections of inert protein, but note that M20 mice develop neither synuclein pathology nor neurodegeneration.
Did the surviving dopaminergic neurons in the fibril-injected M20 mice cope with α-synuclein inclusions, or did they not accumulate any? The researchers found it was the latter. The inclusions co-localized with Th-negative neurons and GFAP-positive astrocytes. In non-transgenic mice, inclusions only appeared in non-dopaminergic neurons, not in astrocytes.
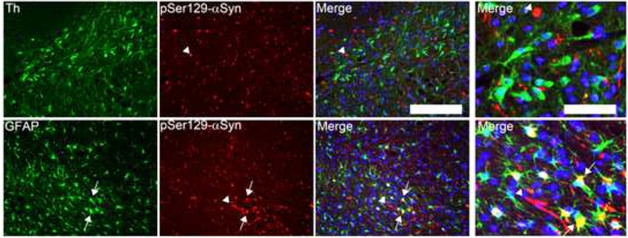
Accumulating Astrocytes. Dopaminergic neurons (green, top panel) appeared devoid of phosphorylated α-synuclein (red), but GFAP+ astrocytes (green, bottom panel) co-localized with the pathology (yellow, right bottom panel). Cell nuclei are labelled with DAPI (blue). [Image courtesy of Sorrentino et al., Molecular Neurodegeneration, 2017.]
In summary, the researchers found that injecting synuclein fibrils into the CPu or IC of M20 mice led to similar distributions of synuclein inclusions, including in regions with no clear neuroanatomical connection to the injection site. In contrast, the inclusions only appeared in regions neuroanatomically connected to the injection site in non-transgenic animals. Because α-synuclein pathology peppered astrocytes of M20 animals, the researchers proposed that α-synuclein pathology also spreads via those cells or even by simple diffusion, but they don’t discount propagation via neural circuitry. “We need to look at synucleinopathies with fresh eyes, and consider the idea that glia may be involved in propagation,” Chakrabarty told Alzforum.
The researchers speculated that different structural conformers or “strains” of α-synuclein might prefer to propagate in astrocytes as opposed to neurons, and that perhaps their particular fibril preparation method produced such an astrocyte-loving strain. Furthermore, that neurons were spared in mice with the most robust gliosis suggests that such neuroinflammatory responses may be beneficial, Chakrabarty told Alzforum. This meshes with her recent finding that stoking neuroinflammation prior to fibril injection prevented proteopathic spread (see Koller et al., 2017).
“This paper raises the tantalizing possibility that astrocytes might play a role in trafficking small amounts of α-synuclein,” commented Kelvin Luk of the University of Pennsylvania in Philadelphia. However, he added that this may simply reflect the fact that M20 mice overexpress the protein in astrocytes, cells that normally do not make the protein. Luk told Alzforum that he also observed astrocytic α-synuclein pathology in A53T α-synuclein transgenic mice. Like Chakrabarty, Luk has not observed synuclein pathology in astrocytes of non-transgenic animals.
Dennis Selkoe of Brigham and Women’s Hospital in Boston cautioned that the approach of injecting preformed fibrils is far from physiological (as reviewed in Walsh and Selkoe, 2016). He added, “We see little evidence that astrocytes accumulate α-synuclein abnormally in Parkinson’s disease and dementia with Lewy bodies, so these findings may be further evidence that the experimental approach being used here is not mimicking those two human diseases.” Giasson and Chakrabarty note that some synucleinopathies, including multiple system atrophy, do feature prominent astrocytic pathology.
Lary Walker of Emory University in Atlanta commented that the astrocytic accumulation does not contradict the predominance of interneuronal transfer. Perhaps astrocytes gobble up fibrils locally, after they have spread to different regions via neurons. Walker and Luk both pointed out that because the authors only considered one time point, it is difficult to rule out all neuroanatomical routes of propagation.
Walker said the unique phenotypes the authors observed—namely little neuronal death and α-synuclein accumulation in non-neuronal cells—underscores a need to understand why publications in this line of research contradict each other. Variations in fibril preparation protocols and mouse strains may be partly to blame. —Jessica Shugart
References
News Citations
- Synthetic Synuclein Corrupts Native Along Mouse Brain Networks
- Toxic Synuclein Corrupts Native in Wild-Type Mice
- Shape of α-Synuclein Aggregates Influences Pathology
- Lewy Pathology in DLB Spreads Fast, Maybe From the Nose
- Are Synuclein Seeds Non-Starters?
- Presenting: A Not-Quite-Parkinson's Mouse Model
Paper Citations
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Kubo M, Shimozawa A, Akiyama H, Hasegawa M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol Commun. 2014 Aug 6;2:88. PubMed.
- Paumier KL, Luk KC, Manfredsson FP, Kanaan NM, Lipton JW, Collier TJ, Steece-Collier K, Kemp CJ, Celano S, Schulz E, Sandoval IM, Fleming S, Dirr E, Polinski NK, Trojanowski JQ, Lee VM, Sortwell CE. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis. 2015 Oct;82:185-99. Epub 2015 Jun 17 PubMed.
- Breid S, Bernis ME, Babila JT, Garza MC, Wille H, Tamgüney G. Neuroinvasion of α-Synuclein Prionoids after Intraperitoneal and Intraglossal Inoculation. J Virol. 2016 Oct 15;90(20):9182-93. Print 2016 Oct 15 PubMed.
- Shimozawa A, Ono M, Takahara D, Tarutani A, Imura S, Masuda-Suzukake M, Higuchi M, Yanai K, Hisanaga SI, Hasegawa M. Propagation of pathological α-synuclein in marmoset brain. Acta Neuropathol Commun. 2017 Feb 2;5(1):12. PubMed.
- Koller EJ, Brooks MM, Golde TE, Giasson BI, Chakrabarty P. Inflammatory pre-conditioning restricts the seeded induction of α-synuclein pathology in wild type mice. Mol Neurodegener. 2017 Jan 3;12(1):1. PubMed.
- Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci. 2016 Apr;17(4):251-60. PubMed.
Further Reading
Primary Papers
- Sorrentino ZA, Brooks MM, Hudson V 3rd, Rutherford NJ, Golde TE, Giasson BI, Chakrabarty P. Intrastriatal injection of α-synuclein can lead to widespread synucleinopathy independent of neuroanatomic connectivity. Mol Neurodegener. 2017 May 29;12(1):40. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.