Are Upper Motor Neuron Gaffes a Prelude to Disease?
Quick Links
Corticospinal motor neurons are the motor system’s foreman, according to Hande Özdinler of the Northwestern University Feinberg School of Medicine in Chicago. They listen to what the brain wants, and then tell lower motor neurons to do it. If the foreman relays the wrong message, movement disorders such as amyotrophic lateral sclerosis or Parkinson’s disease can ensue. In the January 16 Cerebral Cortex online, Özdinler and colleagues show how corticospinal motor neurons (CSMN) are exquisitely sensitive to cellular stress. When stressed, their dendrites degenerate, effectively blocking incoming signals and garbling the instructions they transmit. In the same online issue, scientists from the University of Rome report that the CSMN communicate too much in a mouse model of ALS, firing rapidly and releasing streams of potentially toxic glutamate, which may contribute to their demise.

Dying Dendrites.
In mice lacking UCHL1 (right), the dendrites of corticospinal motor neurons wither. [Image courtesy of Özdinler lab, Northwestern University Feinberg School of Medicine.]
Garbage in …
In Özdinler’s lab, joint first authors Javier Jara and Barış Genç were intrigued by reports that movement disorders result from mutations in the gene ubiquitin carboxy-terminal hydrolase L1 (UCHL1). One study found a variant in the gene in brothers with Parkinson’s disease; another reported a UCHL1 mutation in siblings with a childhood-onset disorder including ataxia and spasticity (Leroy et al., 1998; Bilguvar et al., 2013). While a meta analysis supported UCHLI as a PD gene, not all subsequent studies confirmed that (Maraganore et al., 2004; Wintermeyer et al., 2000; Lincoln et al., 1999).
Regardless of the controversy, Özdinler saw a clue that UCHL1 could be important for the biology of CSMN. UCHL1 contributes to protein homeostasis by both adding ubiquitin to and removing it from other proteins.
Jara, Genç, and colleagues obtained mice with mutant, nonfunctional UCHL1 (Walters et al., 2008). These animals’ limbs turn stiff as early as 40 days of age, and they can barely walk by 65 days. Examining their brains, Jara saw that the CSMN began degenerating by 40 days (see image above). By 100 days, most of those neurons had disappeared, and the ones that remained were shrunken, with sparse dendritic arbors. However, nearby non-motor neurons were fine, suggesting the CSMN, in particular, were susceptible to UCHL1 loss.
What makes them so sensitive? Because UCHL1 contributes to protein quality control, much of which takes place in the endoplasmic reticulum, Özdinler and colleagues hypothesized that the CSMN might face severe ER stress. Sure enough, they observed an excess of ubiquitinated proteins and ER stress markers in the CSMN of UCHL1-null mice. In cell culture, even wild-type CSMN died in the presence of tunicamycin or bortezomib, inducers of ER stress, while other neural types survived.
… Garbage Out
The researchers in Rome, led by Cristina Zona, examined how defects, including dendrite degeneration, altered the output of CSMN. Joint first authors Maria Teresa Viscomi and L. Saba used electrophysiology to examine the signaling of cortical motor neurons in ALS model mice that overexpress a mutant superoxide dismutase 1 gene. The motor neurons Zona examined may or may not be the exact same CSMN studied in Özdinler's group. Viscomi and Saba focused on pyramid-shaped neurons in layer V of the cortex, which fired in a repeating pattern akin to that of CSMN. This suggests, but does not confirm, that the neurons were CSMN, commented Özdinler, who prefers to use retrograde labeling or other specific markers to identify CSMN.
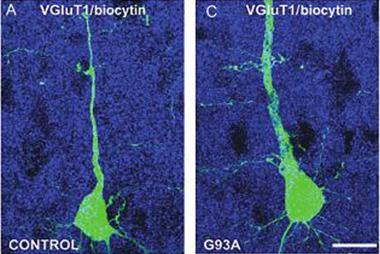
Too Much of a Good Thing.
ALS mice (right) make more of the glutamate transporter vGluT2 (red) than wild-type controls, which could cause excitotoxicity. [Image courtesy of Saba et al., cercor.oxfordjournals.org.]
Nevertheless, in brain slices, the purported mSOD1 CSMN fired more frequently, either spontaneously or in response to stimulation, than control neurons from wild-type mice. What might cause this hyperexcitability? The authors reasoned that the presynaptic vesicles might be overloaded with glutamate. Because the vesicular glutamate transporters vGluT1 and vGluT2 are responsible for filling those vesicles, the authors examined their expression in the motor cortex. The SOD1 mice had normal vGluT1 levels, but more vGluT2 (see image at left). This could enhance glutamate release, which would be toxic to the neurons, the authors surmised. “These alterations indicate that the cortical area has a fundamental role in ALS disease onset and progression and should thus be taken into account for treatment of this neurodegenerative disease,” they wrote.
Nigel Leigh of Brighton and Sussex Medical School in the United Kingdom praised the contributions of these papers, but said it was difficult to link them directly to the pathology of a specific disease such as ALS. That link may come out of future studies. For example, Leigh suggested, geneticists might want to look for mutations in UCHL1 or related genes in people with ALS. He was also curious how the observed defects affect lower motor neurons, which, by definition, are also involved in ALS.
These papers underscore that upper motor neurons contribute to ALS, commented Matthew Fogarty of the University of Queensland in St. Lucia, Australia. “It would be prudent not to overlook the importance of the upper motor neurons … if we are to improve translational outcomes,” he wrote to Alzforum, adding that he thinks both UCHL1 and glutamate excitotoxicity have the potential to be important factors in ALS.
What about other diseases? “In many, the brunt falls upon the upper motor neuron,” said Leigh. Hereditary spastic paraplegia is the classic example, but Parkinson’s, primary lateral sclerosis, and spinal cord injuries might also respond to treatments that target CSMN neurons. Özdinler proposed that UCHL1-negative mice could be a model for preclinical studies.—Amber Dance
References
Paper Citations
- Leroy E, Boyer R, Auburger G, Leube B, Ulm G, Mezey E, Harta G, Brownstein MJ, Jonnalagada S, Chernova T, Dehejia A, Lavedan C, Gasser T, Steinbach PJ, Wilkinson KD, Polymeropoulos MH. The ubiquitin pathway in Parkinson's disease. Nature. 1998 Oct 1;395(6701):451-2. PubMed.
- Bilguvar K, Tyagi NK, Ozkara C, Tuysuz B, Bakircioglu M, Choi M, Delil S, Caglayan AO, Baranoski JF, Erturk O, Yalcinkaya C, Karacorlu M, Dincer A, Johnson MH, Mane S, Chandra SS, Louvi A, Boggon TJ, Lifton RP, Horwich AL, Gunel M. Recessive loss of function of the neuronal ubiquitin hydrolase UCHL1 leads to early-onset progressive neurodegeneration. Proc Natl Acad Sci U S A. 2013 Feb 26;110(9):3489-94. Epub 2013 Jan 28 PubMed.
- Maraganore DM, Lesnick TG, Elbaz A, Chartier-Harlin MC, Gasser T, Krüger R, Hattori N, Mellick GD, Quattrone A, Satoh J, Toda T, Wang J, Ioannidis JP, de Andrade M, Rocca WA, . UCHL1 is a Parkinson's disease susceptibility gene. Ann Neurol. 2004 Apr;55(4):512-21. PubMed.
- Wintermeyer P, Krüger R, Kuhn W, Müller T, Woitalla D, Berg D, Becker G, Leroy E, Polymeropoulos M, Berger K, Przuntek H, Schöls L, Epplen JT, Riess O. Mutation analysis and association studies of the UCHL1 gene in German Parkinson's disease patients. Neuroreport. 2000 Jul 14;11(10):2079-82. PubMed.
- Lincoln S, Vaughan J, Wood N, Baker M, Adamson J, Gwinn-Hardy K, Lynch T, Hardy J, Farrer M. Low frequency of pathogenic mutations in the ubiquitin carboxy-terminal hydrolase gene in familial Parkinson's disease. Neuroreport. 1999 Feb 5;10(2):427-9. PubMed.
- Walters BJ, Campbell SL, Chen PC, Taylor AP, Schroeder DG, Dobrunz LE, Artavanis-Tsakonas K, Ploegh HL, Wilson JA, Cox GA, Wilson SM. Differential effects of Usp14 and Uch-L1 on the ubiquitin proteasome system and synaptic activity. Mol Cell Neurosci. 2008 Dec;39(4):539-48. Epub 2008 Aug 15 PubMed.
Further Reading
Papers
- Jara JH, Villa SR, Khan NA, Bohn MC, Ozdinler PH. AAV2 mediated retrograde transduction of corticospinal motor neurons reveals initial and selective apical dendrite degeneration in ALS. Neurobiol Dis. 2012 Aug;47(2):174-83. PubMed.
- Jiang HQ, Ren M, Jiang HZ, Wang J, Zhang J, Yin X, Wang SY, Qi Y, Wang XD, Feng HL. Guanabenz delays the onset of disease symptoms, extends lifespan, improves motor performance and attenuates motor neuron loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neuroscience. 2014 Sep 26;277:132-8. Epub 2014 Mar 31 PubMed.
- Wang L, Popko B, Tixier E, Roos RP. Guanabenz, which enhances the unfolded protein response, ameliorates mutant SOD1-induced amyotrophic lateral sclerosis. Neurobiol Dis. 2014 Nov;71:317-24. Epub 2014 Aug 15 PubMed.
- Genç B, Özdinler PH. Moving forward in clinical trials for ALS: motor neurons lead the way please. Drug Discov Today. 2014 Apr;19(4):441-9. Epub 2013 Oct 27 PubMed.
- Novarino G, Fenstermaker AG, Zaki MS, Hofree M, Silhavy JL, Heiberg AD, Abdellateef M, Rosti B, Scott E, Mansour L, Masri A, Kayserili H, Al-Aama JY, Abdel-Salam GM, Karminejad A, Kara M, Kara B, Bozorgmehri B, Ben-Omran T, Mojahedi F, Mahmoud IG, Bouslam N, Bouhouche A, Benomar A, Hanein S, Raymond L, Forlani S, Mascaro M, Selim L, Shehata N, Al-Allawi N, Bindu PS, Azam M, Gunel M, Caglayan A, Bilguvar K, Tolun A, Issa MY, Schroth J, Spencer EG, Rosti RO, Akizu N, Vaux KK, Johansen A, Koh AA, Megahed H, Durr A, Brice A, Stevanin G, Gabriel SB, Ideker T, Gleeson JG. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014 Jan 31;343(6170):506-11. PubMed.
- Foran E, Bogush A, Goffredo M, Roncaglia P, Gustincich S, Pasinelli P, Trotti D. Motor neuron impairment mediated by a sumoylated fragment of the glial glutamate transporter EAAT2. Glia. 2011 Nov;59(11):1719-31. PubMed.
- Foran E, Trotti D. Glutamate transporters and the excitotoxic path to motor neuron degeneration in amyotrophic lateral sclerosis. Antioxid Redox Signal. 2009 Jul;11(7):1587-602. PubMed.
- Spalloni A, Nutini M, Longone P. Role of the N-methyl-d-aspartate receptors complex in amyotrophic lateral sclerosis. Biochim Biophys Acta. 2013 Feb;1832(2):312-22. PubMed.
News
Primary Papers
- Jara JH, Genç B, Cox GA, Bohn MC, Roos RP, Macklis JD, Ulupınar E, Özdinler PH. Corticospinal Motor Neurons Are Susceptible to Increased ER Stress and Display Profound Degeneration in the Absence of UCHL1 Function. Cereb Cortex. 2015 Jan 16; PubMed.
- Saba L, Viscomi MT, Caioli S, Pignataro A, Bisicchia E, Pieri M, Molinari M, Ammassari-Teule M, Zona C. Altered Functionality, Morphology, and Vesicular Glutamate Transporter Expression of Cortical Motor Neurons from a Presymptomatic Mouse Model of Amyotrophic Lateral Sclerosis. Cereb Cortex. 2015 Jan 16; PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.