Amyloids Fibrillize, or Stay Solo, Based on Liaisons with Like Proteins
Quick Links
Repetitive, uniform structures are a hallmark of most amyloid fibrils. Alas, despite their apparent uniformity, amyloids do not form in a vacuum. Rather, entanglements with myriad other proteins play a part in their creation. Two studies investigated these so-called heterotypic interactions, and how they might fuel—or put the kibosh on—amyloid formation. One, published November 29 in EMBO and led by Frederic Rousseau and Joost Schymkowitz of KU Leuven, Belgium, identified thousands of proteins with sequences that resemble aggregation-prone regions of Aβ. It reports that some of these proteins not only accelerate Aβ aggregation in vitro, but were also seen mingling with Aβ plaques in postmortem brains. A second study, posted on bioRxiv and led by the same authors, explored the thermodynamic rules of engagement between a sequence at the heart of tau fibrils—VQIVYK—and other proteins bearing a similar sequence. The authors identified proteins that either promote or hinder tau's aggregation, and leveraged their insights to concoct an aggregation capper that thwarted tau fibrillization.
- Thousands of proteins bear sequences that strongly resemble aggregation-prone regions of amyloids.
- APR-like sequences interact with amyloidogenic proteins, boosting or halting their aggregation.
- An aggregation-blocking peptide caps tau fibrils.
The findings suggest that third-party proteins play a meaningful part in proteopathic aggregation, and that their expression patterns could dictate the vulnerability of some cell types to amyloid formation.
At the heart of every type of amyloid fibril are core sequences that are necessary for fibril formation. At five to 15 residues in length, these aggregation-prone regions (APRs) have a high propensity to snap into the cross-β-sheet structures characteristic of amyloids. These short segments can form stable amyloids on their own, or as part of a full-length protein. While APRs tend to coax amyloid formation by teaming up with their own kind, could other proteins bearing homologous APR sequences also set off a fibrillization cascade? This was the main question that preoccupied first author Katerina Konstantoulea and colleagues.

Universal Tau Fibril Capper? CAP1 aggregation blocker (blue and red) designed based on thermodynamic modeling integrated with cryo-EM structures of tau fibrils from, clockwise, CTE, Pick's, CBD, Alzheimer's disease. [Courtesy of Louros et al., bioRxiv, 2021.]
In one study, they focused on the Aβ42 peptide. Aβ42 contains two APRs. One, encompassing residues 16-21, is where several familial AD mutations cluster. The other resides at the peptide's C-terminus. First, the researchers confirmed that these two APRs interact with, and instigate fibrillization of, full-length Aβ42 peptides. Indeed, by passing monomeric and aggregated forms of Aβ over membranes dotted with different 12-residue sequences spanning the length of the peptide, they found that Aβ42 preferentially bound to the APRs.
Would third-party proteins containing sequences similar to Aβ's APRs also ensnare Aβ42? The scientists first asked how many proteins contain such sequences. They scanned the human proteome for sequences containing the core six amino acids in each Aβ APR and, lo and behold, found two proteins containing an identical match, 61 with one mismatch, and 4,318 containing a sequence with two mismatches with an Aβ APR. From this crop of potential Aβ interactors, the researchers tested 600, including their flanking sequences, for interaction with Aβ42 oligomers. This identified 126 protein sequences that latched onto Aβ APRs. In further experiments with a subset of these, the researchers identified several that indeed sped up Aβ42 aggregation in solution.
If proteins containing Aβ-APR-like sequences were to contribute to amyloid formation in situ, then one would expect to find them mingling with Aβ plaques in the AD brain, the researchers reasoned. They combed through published proteomic data from Aβ plaques in the hippocampi of people who had died with AD (Xiong et al., 2019). Compared to the proteomes of nearby tissue, the plaque proteomes were highly enriched for third-party proteins containing sequences resembling those within the Aβ peptide. In particular, these proteins contained sequences with high homology to Aβ APRs.
Interestingly, many of the Aβ plaque-associated proteins containing Aβ-APR-like sequences were involved in synaptic function. Aβ-APR-like sequences were not overrepresented within aggregates of α-synuclein extracted from brain samples of people with multisystem atrophy, suggesting that the Aβ-APR sequences are specifically involved in Aβ aggregation.
The researchers did not find a glut of proteins containing such sequences within amyloid plaques in APP/PS1 mice. This suggested that in this transgenic model—which overexpresses APP—such heterotypic interactions do not influence amyloidosis.
Finally, the researchers picked 10 Aβ-APR-containing proteins they had identified in the proteome, including five that showed up in Aβ plaques, for further analysis in cell culture. To gauge how each protein might sway Aβ aggregation kinetics, they overexpressed them in Aβ biosensor cell lines. Adding Aβ seeds to these cells instigates aggregation of fluorescently tagged Aβ, generating fluorescent puncta. When overexpressed, three of the proteins—CPT2, PLAPG6, and CLNCN3—boosted Aβ aggregation. This was abolished when the researchers mutated the APR-like sequences within the proteins, supporting the idea that these proteins enhanced Aβ aggregation by direct interactions with these homologous domains.
Why are do some APR-like sequences speed up aggregation, while others don't affect it or bring it to a halt? As Schymkowitz put it, “Is there some sort of grammar for amyloidosis?” First author Nikolaos Louros and colleagues addressed this question in the next study, which posted on bioRxiv July 30. They compiled 83 APR sequences from across 18 amyloidogenic proteins, including Aβ, tau, and α-synuclein. Using computational methods, they systematically changed different amino acids within each of these APRs and asked how each mutation would influence the propensity of that APR variant to aggregate with its original form. Ultimately, this analysis yielded a rule book of amyloidosis, highlighting specific biochemical interactions that promote rather than hinder aggregation.
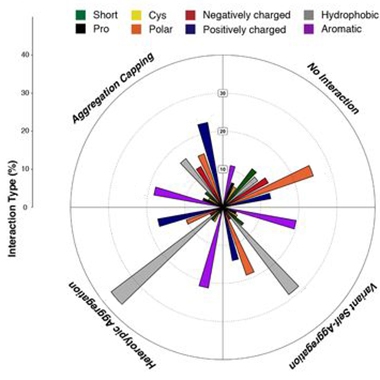
Amyloidosis Grammar. Depending on their biochemical properties (color-coded), different types of amino acid substitution in aggregation-prone regions influenced amyloidosis in different ways. They either capped aggregation, had no interaction, aggregated with themselves, or promoted a heterotypic aggregation with the original APR. [Courtesy of Louros et al., bioRxiv, 2021.]
Firstly, the scientists found that most changes to the sequences of APRs sapped their amyloid-promoting power, supporting the idea that amyloids assemble best with their own kind. Among the APR variants that altered the rate of aggregation, the scientists identified thermodynamic trends associated with aggregation boosters versus blockers. For example, they found that aromatic residues could efficiently cap fibril ends by introducing steric clashes during elongation. Conversely, hydrophobic residues within the core of the APR revved amyloid formation.
Such parameters in place, the researchers tested how well they would hold up against an infamous amyloidogenic protein: tau. The microtubule-binding protein has a well-characterized APR—VQIVYK—which resides within a repeat domain at the C-terminus. The researchers synthesized 90 peptide variants of the sequence, each with different amino acid substitutions, and mixed them with tau. Using thioflavin S to monitor aggregation, the researchers found remarkable agreement between their thermodynamic modeling and reality. For example, peptides with aromatic substitutions in key positions effectively capped tau aggregation, while some variants with hydrophobic cores accelerated it.
Akin to what they had done with Aβ42, the researchers scanned the proteome for proteins containing sequences that were similar to the VQIVYK hexapeptide. They overexpressed 11 of their finds in tau biosensor cell lines. After adding tau seeds to the cells, six of the proteins—IDE, TRA2B, SNTG1, HSPA2, MAPK8IP3, and DOCK3—were spotted mingling within tau inclusions. These proteins significantly accelerated tau aggregation, rendering cells more vulnerable to tau seeding and ramping up the number of tau inclusions per cell.
Finally, the researchers used their findings to design an ideal tau aggregation-blocking peptide. Called CAP1, it consisted of two copies of V1W, a variant of the tau hexapeptide sequence that contained fibril-capping aromatic residues. They found that CAP1 effectively thwarted aggregation of the VQIVYK hexapeptide as well as full-length tau in vitro, and also shut down tau aggregation within biosensor cell lines. The authors claim that CAP1 was five times more potent at halting tau aggregation than previously reported aggregation blockers (Seidler et al., 2018).
In all, the findings imply that each cell’s unique proteome influences that cell’s vulnerability to amyloid aggregation. “The solubility of amyloidogenic proteins can be modulated by specific interactors in their environment,” Rousseau said. They also support the idea that, rather than randomly glomming onto other proteins, amyloids interact based on distinct biochemical rules. “Amyloidogenic proteins can be sticky, but they are sticky in a specific manner,” Rousseau said. —Jessica Shugart
References
Paper Citations
- Xiong F, Ge W, Ma C. Quantitative proteomics reveals distinct composition of amyloid plaques in Alzheimer's disease. Alzheimers Dement. 2019 Mar;15(3):429-440. Epub 2019 Jan 2 PubMed.
- Seidler PM, Boyer DR, Rodriguez JA, Sawaya MR, Cascio D, Murray K, Gonen T, Eisenberg DS. Structure-based inhibitors of tau aggregation. Nat Chem. 2018 Feb;10(2):170-176. Epub 2017 Nov 20 PubMed.
Further Reading
No Available Further Reading
Primary Papers
- Konstantoulea K, Guerreiro P, Ramakers M, Louros N, Aubrey LD, Houben B, Michiels E, De Vleeschouwer M, Lampi Y, Ribeiro LF, de Wit J, Xue WF, Schymkowitz J, Rousseau F. Heterotypic Amyloid β interactions facilitate amyloid assembly and modify amyloid structure. EMBO J. 2022 Dec 17;41(2):e108591. Epub 2021 Nov 29 PubMed.
- Louros N, Ramakers M, Michiels E, Konstantoulea K, Morelli C, Garcia T, D’Haeyer S, Goossens V, Audenaert D, Rousseau F, Schymkowitz J. Mapping the sequence specificity of heterotypic amyloid interactions enables the identification of aggregation modifiers. bioRxiv, July 30, 2021 bioRxiv.
Annotate
To make an annotation you must Login or Register.
Comments
University of Melbourne
Florey Instotute of Neurosciece and Mental Health
Utilizing extensive proteomic data of Aβ plaques from AD patients and multiple sequence alignments, this sound work produced a library of peptides that modify Aβ assembly kinetics, fibril morphology, and deposition pattern, all in vitro. Proteomic analysis showed that enriched proteins harbor homologous sequences to the Aβ aggregation-prone regions (APRs) as defined a priori as 16-21 and the c-terminal 29-end.
It is not surprising that highly hydrophobic “sticky” Aβ aggregates can collect hydrophobic fragments of other proteins through heterotypic interactions. This is another example of how sensitive aggregation of Aβ (or any intrinsically disordered protein) is to variations in conditions: pH, metal binding, and various co-factors. The varying elements are infinite. Modified aggregation produces different forms of Aβ which may have different toxicities and thus may even stabilize nontoxic forms.
Further, in the following publication, the authors have performed a systematic thermodynamic evaluation, coupled with multidimensionality analysis to investigate structural compatibility for the entire sequence space of single variants of the APR dataset. The results “indicated that even for highly conserved sequences, such as single position variants, a thermodynamically favourable fit within the defined aggregation core is rather hardly accommodated. … This apparent incompatibility of APR cores to sequence variation was also supported by the fact that only a limited fraction of sequence variants was predicted to favour self-assembly (6.9%) possibly suggesting that the template backbone arrangements are strongly tailored to their particular sequences.”
Indeed, those sequences are from globular proteins where the “sequence-structures” unique relationship is held. Intrinsically disordered proteins (Aβ, tau, etc.) challenge this postulate. A further support to this observation could be our test run of DeepMind Alphafold2 AI system for predicting Aβ or tau folds. The system iterates between multiple sequence alignment (as above) plus residue pairs to predict pairwise distances between all protein residues. The selected sequences can form potentially an additional library of peptides to be analyzed and tested. However, the system fails to predict any stable sensible structural templates/folds as expected.
With so many variables in play, it is difficult to interpret the biological significance of the findings. The best guideposts still seem to be the hits thrown up by genetic analyses.
Make a Comment
To make a comment you must login or register.