ALS on a Chip—A Model for Screening Therapeutics?
Quick Links
Though motor neurons are the primary victims in amyotrophic lateral sclerosis, it is their waning connections with muscles that ultimately triggers symptoms of the disease. This complex interaction can now be mimicked on a tiny silicone chip, according to a paper in Science Advances on October 10. Researchers led by Roger Kamm of the Massachusetts Institute of Technology derived both muscle fibers and motor neurons from human stem cells, placed them into adjacent microfluidic chambers etched onto a polymer surface, and watched as motor neurons extended axons to form functional junctions with the muscle cells, even triggering their contraction. Tellingly, when those motor neurons came from an ALS patient, they formed weaker connections, the muscles contracted poorly, and they eventually withered. The researchers reported that treatment with small molecules rescued the motor deficits. The microfluidic device could therefore prove useful for screening therapeutics, the researchers contend.
- Functional neuromuscular junctions formed between muscle fibers and motor neurons in a microfluidic device.
- When motor neurons came from an ALS patient, junctions performed poorly, and muscle fibers degenerated.
- Treatment with bosutinib and rapamycin rescued deficits.
“ALS-on-a-chip is a brilliant model that might facilitate a better understanding of the complex relationship between motor neurons and muscle cells,” wrote Richard Bedlack of Duke University in a comment to Alzforum. “It might even give us a simple way to screen large numbers of drugs at a fraction of the cost and time required using more traditional animal models.”
Efforts to develop a disease-modifying therapy for ALS have been hampered by the lack of models that closely mimic the complex dynamics between motor neurons and muscle fibers. In an attempt to recreate these neuromuscular junctions, also known as motor units, several labs have turned to “organ-on-a-chip” technology, in which different cell types (in this case, motor neurons and muscle cells) are placed into neighboring chambers separated by collagen or tiny tunnels (Southam et al., 2013; Morimoto et al., 2013; Santhanam et al., 2018). Axons can then extend from the neuronal chamber to the muscle chamber, where they form neuromuscular junctions and trigger contractions. Kamm previously used embryonic stem cells derived from mice to generate muscle fibers and motor neurons (Uzel et al., 2016).
For the current study, first author Tatsuya Osaki and colleagues used human cells. To generate muscle fibers, they put iPSC-derived skeletal muscle cells into a chamber containing small flexible pillars, around which the cells spontaneously formed muscle fiber bundles. The researchers could measure the deflection of these pillars to gauge the force of muscle contractions. They also coaxed human embryonic stem cells (hESCs) to form motor neurons and astrocytes. They transduced the developing cells with the gene for channelrhodopsin-2, a light-sensitive ion channel, which allowed them to orchestrate synchronized action potentials in the motor neurons. Once the motor neurons matured, the researchers placed them into the chamber adjacent to the muscle fiber compartment.
The motor neurons sprouted axons that grew through the collagen layer separating the two chambers. By four days, they had reached the muscle fibers and had formed neuromuscular junctions. By 14 days, the nerve fibers had thickened and the junctions stimulated random contractions in the muscle cells. The researchers could synchronize these contractions optogenetically by activating the channelrhodopsin or by adding neurotransmitters.
To investigate whether neuromuscular junction deficits that occur in ALS could be modeled within their chip, the researchers generated motor neurons from iPSCs derived from a patient diagnosed with sporadic ALS. Subsequent whole exome sequencing and genotyping revealed that the cells harbored a pathogenic mutation in TDP-43, and the derived motor neurons had cytoplasmic aggregates of the protein.
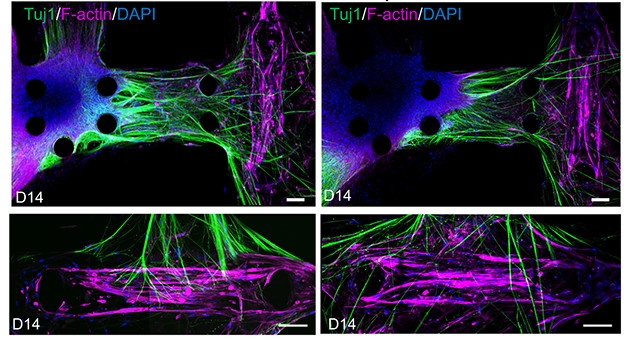
Missed Connections. In motor units derived from a healthy control (left panels), nerve fibers (green) project from motor neurons (blue) to muscle fibers (pink). Units derived from an ALS patient are less robust (right panels). [Courtesy of Osaki et al., Science Advances, 2018.]
Compared with motor neurons from a healthy control, the ALS motor neurons took several days longer to connect to muscle fibers, and ultimately they formed fewer thick nerve fibers. Stimulating them with light triggered action potentials only about 60 percent of the time, compared with nearly 100 percent responsiveness of non-ALS motor neurons. By 14 days in culture, muscle contractions triggered by the ALS motor neurons were about a quarter as strong as those triggered by non-ALS motor neurons. Levels of apoptotic markers had increased in the muscle cells, suggesting they had begun to die off.
To investigate if the junctions formed by ALS-derived cells could be strengthened, the researchers added the autophagy inducer rapamycin, either alone or in combination with bosutinib—an inhibitor of the Scr/c-Abl pathway. Rapamycin is currently being tested in a Phase 2 clinical trial for ALS, while bosutinib is being investigated in a Phase 1 trial for cognitive impairment (see, respectively, clinicaltrials.gov; clinicltrials.gov). The drugs improved the responsiveness of the motor neurons as well as the strength of muscle contractions, and dampened the expression of apoptotic markers. While rapamycin alone offered partial benefits for these measures, the combination of both drugs worked best. Notably, the drugs stimulated autophagy and reduced the accumulation of TDP-43 in the motor neuron cytoplasm.
Kamm told Alzforum that the researchers are partnering with Biogen in Cambridge, Massachusetts, to further optimize the chip for screening therapeutics. They have also generated a version of the chip with a layer of endothelial cells separating the two chambers, in an effort to mimic the blood-brain barrier.
James Hickman of the University of Central Florida in Orlando called the study a solid step toward modeling an ALS neuromuscular junction on a chip. Hickman previously developed a very similar model, albeit with motor neurons from healthy controls. In his model, motor neurons were stimulated with electrical currents, and axon-sized microtunnels connected the chambers, instead of a layer of collagen. He told Alzforum that the strict separation of the two chambers with microtunnels was critical for testing the efficacy of drugs aimed specifically at motor neurons because this method prevents drugs from diffusing into the adjacent chamber. However, he said Kamm’s model could prove useful for early stage screening.
Hickman also noted that given the small number of motor neuron samples used—one from a control, and one from an ALS patient—it is difficult to conclude whether differences in the function of neuromuscular junctions were actually a result of the disease process, or just experimental variability. Genetic variation beyond the TDP-43 mutation could also contribute to the differences, he said. Kamm told Alzforum that this initial study was a proof-of-concept, and that he plans to extend it to motor neurons from many more patients. To eliminate the confounds of genetic background, the researchers will also test isogenic lines, using gene editing to correct pathogenic mutations in control cells.—Jessica Shugart
References
Paper Citations
- Southam KA, King AE, Blizzard CA, McCormack GH, Dickson TC. Microfluidic primary culture model of the lower motor neuron-neuromuscular junction circuit. J Neurosci Methods. 2013 Sep 15;218(2):164-9. Epub 2013 Jun 14 PubMed.
- Morimoto Y, Kato-Negishi M, Onoe H, Takeuchi S. Three-dimensional neuron-muscle constructs with neuromuscular junctions. Biomaterials. 2013 Dec;34(37):9413-9. Epub 2013 Sep 14 PubMed.
- Santhanam N, Kumanchik L, Guo X, Sommerhage F, Cai Y, Jackson M, Martin C, Saad G, McAleer CW, Wang Y, Lavado A, Long CJ, Hickman JJ. Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials. 2018 Jun;166:64-78. Epub 2018 Feb 27 PubMed.
- Uzel SG, Platt RJ, Subramanian V, Pearl TM, Rowlands CJ, Chan V, Boyer LA, So PT, Kamm RD. Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci Adv. 2016 Aug;2(8):e1501429. Epub 2016 Aug 3 PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Osaki T, Uzel SG, Kamm RD. Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci Adv. 2018 Oct;4(10):eaat5847. Epub 2018 Oct 10 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.