ABAD, aka ERAB: Mitochondrial Miscreant Returns
Quick Links
The Aβ-binding alcohol dehydrogenase (ABAD) forms a complex with Aβ in mitochondria, promoting leakage of free radicals, mitochondrial dysfunction, and cell death, according to a combined structure/function and animal study to be published in Science today. The paper proposes a molecular mechanism for how Aβ kills neurons; the current absence of a consensus mechanism for Aβ toxicity is one of the most prominent gaps of the amyloid hypothesis.
At the tail end of the last millennium, a spotlight shone briefly on an enzyme that appeared to bind Aβ in the endoplasmic reticulum. Initially dubbed "ER-associated binding protein" (ERAB), it was also found to be elevated in some neurons in AD brain (see Yan (abstr. 881) in Alzforum meeting report.) But then the true site of the enzyme's tryst with Aβ turned out to be mitochondria, and it was renamed ABAD (see, e.g., He et al., 2002). It also carries the moniker 7β-hydroxysteroid dehydrogenase (HSD), type 10 (or just HSD-10), and is known to modulate intracellular levels of sex steroids in non-neural cells (see, e.g., Ivell et al., 2003; Wen et al., 2002).
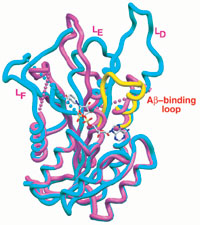
In this figure the Aβ-bound human ABAD complex is shown in pink. For comparison, it is superimposed on the rat ABAD in complex with NAD (blue). The L[D] loop of 3∀-hydroxysteroid dehydrogenase (3∀-HSD) is shown in yellow, and NAD is shown as a stick model in the center. Also shown is the proposed Aβ-binding loop. (Image copyright Science)
Shirley ShiDu Yan of Columbia University in New York City and colleagues have been pursuing this molecule since before its name change and found, among other things, that ABAD might mediate the cellular stress induced by Aβ (Yan et al., 2001). In their most recent study—a collaboration with biochemist Hao Wu's team at Weill Medical College of Cornell University in New York City, as well as with researchers elsewhere—Yan's team explores how and where this potentially destructive coupling takes place.
The researchers first established that there is a significant presence of the ABAD-Aβ complex in human AD brain, as opposed to almost none in normal age-matched controls. The researchers also fingered the complex in mitochondria of 12-month-old mice transgenic for human mutant APP or for both mAPP and ABAD. A closer look with confocal and electron microscopy showed that the ABAD/Aβ complex was taking up residence in the mitochondria of AD-derived cortical brain tissue, as well as in the transgenic mAPP/ABAD mice.
In a further series of experiments, the researchers probed the structural relationships of Aβ and ABAD. They solved the crystal structure of human ABAD in the presence of excess nicotinamide adenine dinucleotide (NAD) and Aβ. (NAD is a cofactor in the enzyme reaction mediated by ABAD.) They determined that Aβ binds to ABAD in such a way as to exclude NAD binding. They also saw that the critical segment of ABAD for recognition of Aβ appears to be the L[D] loop. Armed with this knowledge, the researchers were able to design a selective inhibitor of the Aβ-ABAD interaction, termed the decoy peptide, or ABAD-DP. It consists simply of the short peptide segment that includes the L[D] loop.
Crystal packing of many separate ABAD molecules, with the ordered ends of the L[D] loops (dotted pink lines) pointing into interconnected solvent channels. Each ABAD molecule is shown in a different color. (Image copyright Science)

Using this inhibitor, the researchers demonstrate in cultured neurons that Aβ-induced oxidative stress is dependent upon ABAD. The ABAD-DP prevents the production of reactive oxygen species, DNA fragmentation, and cell death. And finally, what AD tour de force would be complete without a water maze experiment? The researchers tested transgenic mAPP/ABAD in the radial arm water maze and report that their spatial and temporal memory was severely impaired.
"Taken together, these studies establish that Aβ may exert an important pathogenic role in the mitochondrial compartment through an interaction with ABAD, and that inhibition of ABAD-Aβ interaction may provide a new treatment strategy against AD," conclude the authors. The same issue of Science contains a separate study that also implicates the mitochondria in neurodegeneration and cell stress, this time through the new familial parkinsonism gene Pink1 (see ARF related news story).—Hakon Heimer
References
News Citations
Paper Citations
- He XY, Merz G, Yang YZ, Mehta P, Schulz H, Yang SY. Characterization and localization of human type10 17beta-hydroxysteroid dehydrogenase. Eur J Biochem. 2001 Sep;268(18):4899-907. PubMed.
- Ivell R, Balvers M, Anand RJ, Paust HJ, McKinnell C, Sharpe R. Differentiation-dependent expression of 17beta-hydroxysteroid dehydrogenase, type 10, in the rodent testis: effect of aging in Leydig cells. Endocrinology. 2003 Jul;144(7):3130-7. PubMed.
- Wen GY, Yang SY, Kaczmarski W, He XY, Pappas KS. Presence of hydroxysteroid dehydrogenase type 10 in amyloid plaques (APs) of Hsiao's APP-Sw transgenic mouse brains, but absence in APs of Alzheimer's disease brains. Brain Res. 2002 Nov 1;954(1):115-22. PubMed.
- Yan SD, Schmidt AM, Stern D. Alzheimer's disease: inside, outside, upside down. Biochem Soc Symp. 2001;(67):15-22. PubMed.
External Citations
Further Reading
No Available Further Reading
Primary Papers
- Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004 Apr 16;304(5669):448-52. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Weill Medical College of Cornell University
In this article, Lustbader and colleagues investigate what happens when Aβ interacts with ABAD (Aβ-binding alcohol dehydrogenase), the only protein found to interact with Aβ in a yeast two-hybrid screen [1]. In the current work, the authors showed that ABAD and Aβ colocalized to mitochondria by electron microscopy, and could be coimmunoprecipitated from a mitochondrial preparation. Aβ caused the cofactor NAD+ to be excluded from the crystal structure of ABAD, likely explaining the previously observed [2] inhibition of enzymatic activity by Aβ. ABAD levels were increased in pathologically affected areas of AD brain. This is potentially deleterious, because the presence of ABAD exacerbated the cytotoxicity of exogenous Aβ or of expressing a doubly mutated APP, resulting in increased free radical production, cytochrome c release, and DNA fragmentation. Moreover, mice coexpressing ABAD and mutant APP exhibited learning deficits. An ABAD “decoy peptide,” elegantly designed based on the crystal structure, attenuated Aβ-induced cytotoxicity, presumably by preventing the interaction of Aβ and bona fide ABAD.
This is a tantalizing contribution from several points of view. First, it provides a new therapeutic target. Protection from Aβ-induced toxicity was previously obtained by use of an anti-ABAD F(ab')2 [1] and by catalytically inactivating ABAD [2], confirming the results of the ABAD “decoy peptide” approach. The “decoy peptide” has the added advantage that it might prevent other pathologic interactions of Aβ, not just that with ABAD. Second, although the final pathologic hallmarks of Alzheimer’s disease, amyloid plaques, are extracellular, a growing literature suggests that the intracellular accumulation of Aβ may be pathogenetically important [3]. Lustbader and colleagues have suggested a specific molecular target and mechanism through which intracellular Aβ could be toxic. Third, this work links amyloid with the apparently unrelated world of mitochondrial dysfunction and oxidative stress. Mitochondrial dysfunction and oxidative stress are among the earliest events in Alzheimer’s disease [4, 5] and transgenic APP animal models [6], but the mechanisms relating these to Aβ physiology have been unclear.
At the same time, a few cautions are appropriate. A large body of work localizes intracellular APP and Aβ to the secretory or endocytic pathways. Only one prior study suggested that APP could be targeted to mitochondria [7], and Aβ was not mentioned. Thus, the mitochondrial localization of Aβ needs to be confirmed. The rabbit anti-A IgG used to visualize Aβ in mitochondria is not characterized or referenced. Could it cross-react with APP (in agreement with Anandatheerthavarada et al.) or a yet unidentified mitochondrial epitope (does it stain mitochondria in an APP knockout mouse)[7]? Second, there is not yet sufficient evidence to conclude that the pathologic ABAD-Aβ interaction is the one occurring in mitochondria. The authors have previously shown that ABAD is also localized to the ER and, after exposure to Aβ, the inner surface of the plasma membrane [1,2]. The ABAD “decoy peptide” presumably disrupts the ABAD-Aβ interaction at all sites, not just in mitochondria. Thus, toxicity from one of these other sites cannot be excluded. Indeed, in cells coexpressing ABAD and APPV717F, the most intense colocalization of ABAD with hydroxynonenal and malondialydehyde (markers of lipid oxidation) was subplasmalemmal [2].
References:
Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997 Oct 16;389(6652):689-95. PubMed.
Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, Gibson L, Stern E, Collison K, Al-Mohanna F, Ogawa S, Roher A, Clarke SG, Stern DM. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Abeta-induced cytotoxicity. J Biol Chem. 1999 Jan 22;274(4):2145-56. PubMed.
Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's beta-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004 Apr 7;24(14):3592-9. PubMed.
Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA. Mitochondrial abnormalities in Alzheimer's disease. J Neurosci. 2001 May 1;21(9):3017-23. PubMed.
Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001 Aug;60(8):759-67. PubMed.
Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM. Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis. J Neurosci. 2001 Jun 15;21(12):4183-7. PubMed.
Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003 Apr 14;161(1):41-54. PubMed.
Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002 Nov;161(5):1869-79. PubMed.
Yale University School of Medicine
This is an interesting but still incomplete story. This work dates back several years, when the authors found an unexpected ability of the Aβ peptide to bind to the enzyme alcohol dehydrogenase (ADH). It appears that the binding is relatively specific when compared with other peptides, although I am surprised that these peptides don't bind to other proteins non-specifically. The authors had a great opportunity to study the interaction between Aβ and the so-called ABAD protein when they apparently cocrystallized the two. Unfortunately they could not see the Aβ peptide in the complex, so it is impossible to say where Aβ actually binds, or why it blocks the ability of NAD to bind to ABAD.
The paper shows that Aβ and ABAD localize in, around, or next to mitochondria, but not that it is primarily inside the mitochondria, and the immunoelectron microscopy data do not resolve this question.
It is too early to suggest that this latest observation offers therapeutic potential, but one hopes it may with further, more definitive data.
Leeds Trinity University
It has been suggested previously that ERAB (aka ABAD) residues 99-108 contain the Aβ binding domain (Milton et al 2001) so it's nice to see that confirmed using different techniques.
References:
Milton NG, Mayor NP, Rawlinson J. Identification of amyloid-beta binding sites using an antisense peptide approach. Neuroreport. 2001 Aug 8;12(11):2561-6. PubMed.
�
This is the first article I've seen that provides a considerably complete mechanism for the toxicity of β amyloid in Alzheimer's disease. It ties together evidence of Aβ binding to ABAD, mitochondrial stress, and free radical involvement long implicated in Alzheimer's disease.
Are there any suggestions as to how the proposed mechanism may affect synaptic function prior to cell death? (Reference implicating synaptic dysfunction in Alzheimer's is included.)
References:
Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002 Oct 25;298(5594):789-91. PubMed.
Center for Neuroscience and Cell Biology
ABAD—The New/Old Good/Bad Guy in Alzheimer's Disease Lustbader and colleagues [1] present a complex potential mechanism for the role of amyloid β in Alzheimer's disease (AD) pathology. The authors created a crystal form of amyloid β-binding alcohol dehydrogenase (ABAD) and amyloid β that demonstrates that both molecules interact and accumulate inside mitochondria. They suggested that this interaction increases oxidative stress, mitochondrial dysfunction, and cell death occurring in AD.
AD, one of the most devastating age-related neurodegenerative diseases, is associated with oxidative stress, altered energy metabolism, and mitochondrial impairment [1-15]. Postmortem studies revealed a decline in the activities of pyruvate dehydrogenase and alpha-ketoglutarate dehydrogenase [16], key enzymes in energy metabolism that are localized in mitochondria. Furthermore, defects on cytochrome oxidase have also been described [17]. Furthermore, it was demonstrated that Aβ peptides and/or Ca2+ induce the opening of mitochondrial permeability transition pore (a process that results in a nonselective increase in the permeability of the inner mitochondrial membrane to solutes smaller than 1.5 kDa) [18,19]. Moreover, Cardoso et al. [20] showed that amyloid β toxicity requires functional mitochondria.
A previous study [21] implicated endoplasmic reticulum amyloid β-binding protein (ERAB) as a participant in causing neuronal dysfunction in AD. In the same, study the authors observed a strong neuronal ERAB reactivity in the brains of patients with AD, yet it is almost absent in normal brain. Moreover, ERAB is found near amyloid β plaques, and the cellular toxicity of amyloid β can be reduced by blocking ERAB and increased by its overexpression. Furthermore, Oppermann et al. [22] demonstrated that ERAB is localized in the endoplasmic reticulum and mitochondria suggesting a complex interaction with components of the programmed cell death machinery.
ABAD is a member of the family of short chain dehydrogenase/reductase and ERAB has structural homology with short-chain alcohol dehydrogenase (hydroxysteroid dehydrogenases and acetyl-CoA reductases). Furthermore, both appear to potentiate cell stress induced by amyloid β, as evidenced by increased oxidative stress [22, 23]. So, together these findings may suggest that both ABAD and ERAB may interplay in the amyloid β-associated oxidative stress mechanisms or both may be the same entity with two different names. However, these findings suggest a possible target for the development of new therapeutic strategies envisaging the prevention or reduction of the oxidative stress cascade typical of AD pathology.
References:
Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004 Apr 16;304(5669):448-52. PubMed.
Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991 Dec 1;88(23):10540-3. PubMed.
Azari NP, Pettigrew KD, Schapiro MB, Haxby JV, Grady CL, Pietrini P, Salerno JA, Heston LL, Rapoport SI, Horwitz B. Early detection of Alzheimer's disease: a statistical approach using positron emission tomographic data. J Cereb Blood Flow Metab. 1993 May;13(3):438-47. PubMed.
Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994 Nov;36(5):747-51. PubMed.
Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR. Alterations of cerebral metabolism in probable Alzheimer's disease: a preliminary study. Neurobiol Aging. 1994 Jan-Feb;15(1):117-32. PubMed.
Beal MF. Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol. 1995 Sep;38(3):357-66. PubMed.
Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23(1):134-47. PubMed.
Hoyer S. Risk factors for Alzheimer's disease during aging. Impacts of glucose/energy metabolism. J Neural Transm Suppl. 1998;54:187-94. PubMed.
Münch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ, Vlassara H, Smith MA, Perry G, Riederer P. Alzheimer's disease--synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Transm. 1998;105(4-5):439-61. PubMed.
Behl C. Alzheimer's disease and oxidative stress: implications for novel therapeutic approaches. Prog Neurobiol. 1999 Feb;57(3):301-23. PubMed.
Mattson MP, Gary DS, Chan SL, Duan W. Perturbed endoplasmic reticulum function, synaptic apoptosis and the pathogenesis of Alzheimer's disease. Biochem Soc Symp. 2001;(67):151-62. PubMed.
Aliev G, Seyidova D, Lamb BT, Obrenovich ME, Siedlak SL, Vinters HV, Friedland RP, LaManna JC, Smith MA, Perry G. Mitochondria and vascular lesions as a central target for the development of Alzheimer's disease and Alzheimer disease-like pathology in transgenic mice. Neurol Res. 2003 Sep;25(6):665-74. PubMed.
Perry G, Taddeo MA, Nunomura A, Zhu X, Zenteno-Savin T, Drew KL, Shimohama S, Avila J, Castellani RJ, Smith MA. Comparative biology and pathology of oxidative stress in Alzheimer and other neurodegenerative diseases: beyond damage and response. Comp Biochem Physiol C Toxicol Pharmacol. 2002 Dec;133(4):507-13. PubMed.
Su B, Wang X, Nunomura A, Moreira PI, Lee HG, Perry G, Smith MA, Zhu X. Oxidative stress signaling in Alzheimer's disease. Curr Alzheimer Res. 2008 Dec;5(6):525-32. PubMed.
Ghanbari HA, Ghanbari K, Harris PL, Jones PK, Kubat Z, Castellani RJ, Wolozin BL, Smith MA, Perry G. Oxidative damage in cultured human olfactory neurons from Alzheimer's disease patients. Aging Cell. 2004 Feb;3(1):41-4. PubMed.
Blass JP, Sheu RK, Gibson GE. Inherent abnormalities in energy metabolism in Alzheimer disease. Interaction with cerebrovascular compromise. Ann N Y Acad Sci. 2000 Apr;903:204-21. PubMed.
Kish SJ, Bergeron C, Rajput A, Dozic S, Mastrogiacomo F, Chang LJ, Wilson JM, DiStefano LM, Nobrega JN. Brain cytochrome oxidase in Alzheimer's disease. J Neurochem. 1992 Aug;59(2):776-9. PubMed.
Moreira PI, Santos MS, Moreno A, Oliveira C. Amyloid beta-peptide promotes permeability transition pore in brain mitochondria. Biosci Rep. 2001 Dec;21(6):789-800. PubMed.
Moreira PI, Santos MS, Moreno A, Rego AC, Oliveira C. Effect of amyloid beta-peptide on permeability transition pore: a comparative study. J Neurosci Res. 2002 Jul 15;69(2):257-67. PubMed.
Cardoso SM, Santos S, Swerdlow RH, Oliveira CR. Functional mitochondria are required for amyloid beta-mediated neurotoxicity. FASEB J. 2001 Jun;15(8):1439-41. PubMed.
Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D. An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer's disease. Nature. 1997 Oct 16;389(6652):689-95. PubMed.
Oppermann UC, Salim S, Tjernberg LO, Terenius L, Jörnvall H. Binding of amyloid beta-peptide to mitochondrial hydroxyacyl-CoA dehydrogenase (ERAB): regulation of an SDR enzyme activity with implications for apoptosis in Alzheimer's disease. FEBS Lett. 1999 May 28;451(3):238-42. PubMed.
Yan SD, Shi Y, Zhu A, Fu J, Zhu H, Zhu Y, Gibson L, Stern E, Collison K, Al-Mohanna F, Ogawa S, Roher A, Clarke SG, Stern DM. Role of ERAB/L-3-hydroxyacyl-coenzyme A dehydrogenase type II activity in Abeta-induced cytotoxicity. J Biol Chem. 1999 Jan 22;274(4):2145-56. PubMed.
Make a Comment
To make a comment you must login or register.