C9ORF72 Mice A-OK Despite Toxic RNAs, Peptides
Quick Links
Two new mouse models for amyotrophic lateral sclerosis and frontotemporal dementia display classic pathological signs of the C9ORF72 repeats they carry—but no sign of neurodegeneration. The models, generated by the groups of Robert Baloh at the Cedars-Sinai Medical Center in Los Angeles and Robert Brown at the University of Massachusetts Medical School in Worcester, appear side-by-side in the December 2 Neuron. Though scientists were disappointed not to see symptoms reminiscent of ALS or FTD in these animals, they said the models will be useful for testing therapeutics against what the mice do have—abundant RNAs and polypeptides encoded by the repeats.
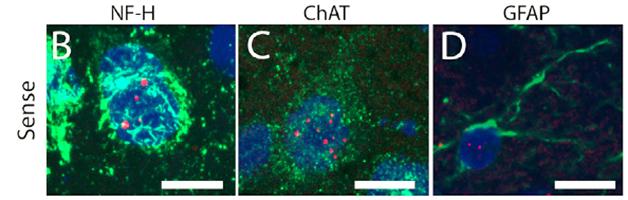
Focus on foci. Clumps of repeat RNA speckle the neurons (B and C) and astrocytes (D) of C9ORF72 model mice. [Neuron, Peters et al.]
Hexanucleotide expansions in the C9ORF72 gene, with repeats numbering in the hundreds or even thousands, are associated with both ALS and FTD. Cells transcribe the repeats in both the sense and antisense directions, and those RNAs form foci that are characteristic of the diseases’ pathology. Furthermore, ribosomes abnormally translate those RNAs, yielding five different poly-dipeptides depending on reading frame (two reading frames produce the same product, poly-glycine-proline). These poly-dipeptides are also standard markers of C9ORF72 pathology.
Several scientists are attempting to copy the human C9ORF72-based disease in mice, and Alzforum first covered Brown’s model earlier this year (see Oct 2015 conference news and table below). Both Brown and Baloh took similar approaches, transferring the repeat-containing C9ORF72 gene from patient DNA to mice by way of a bacterial artificial chromosome. Baloh’s group, in collaboration with Cat Lutz from the Jackson Laboratory in Bar Harbor, Maine, started with a swatch of DNA containing the entire C9ORF72 gene and approximately 800 repeats. At UMass, joint senior authors Brown and Christian Mueller began with a construct containing the first six exons and about 500 repeats.
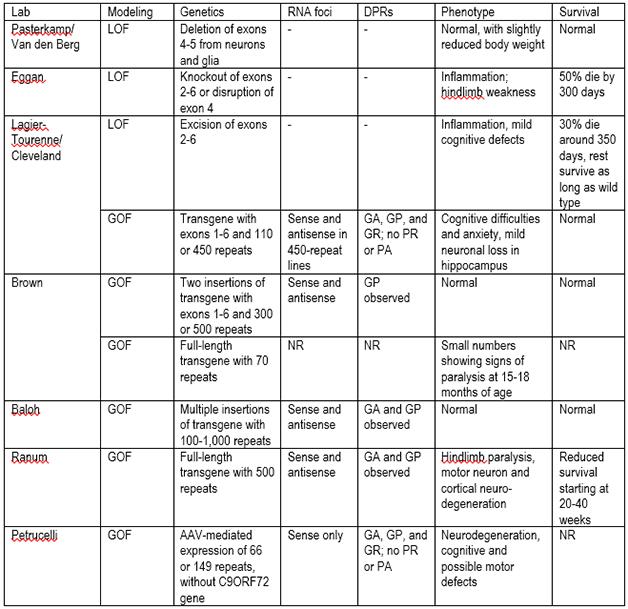
C9ORF72 model mice. LOF=loss of function; GOF=gain of function; DPRs=dipeptide repeats; NR=not reported; GA=glycine-alanine; GP=glycine, proline; GR=glycine-arginine; PR=proline-arginine; PA=proline-alanine.
In bacteria, DNA repeats tend to expand and contract, complicating the BAC approach. In the Brown group, joint first authors Owen Peters and Gabriela Toro Cabrera wound up with mice containing at least two copies of the transgene, one with 300 repeats and one with 500. In Baloh’s, first author Jacqueline O’Rourke obtained dozens of mouse lines with multiple transgene insertions, with repeat lengths from 100 to 1,000. In one bacterial line, the expansion contracted down to 15 repeats, a nonpathogenic number, so the authors used its clone to make a control line. O’Rourke focused her studies on two with the highest levels of transgene expression. These, and the Brown mice, expressed about the same amount of C9ORF72 as human tissues with the expansion, based on quantitative PCR.
All the large expansion lines had obvious C9ORF72-related pathology—both RNA foci and poly-dipeptides. This did not occur in the 15-repeat line. Using available poly-dipeptide antibodies, both groups observed poly-glycine-proline in the brain and spinal cord. It tended to be soluble in young mice, forming more aggregates as the animals aged. Baloh told Alzforum that his group also detected poly-glycine-alanine in the brain, with similar aggregation over time. In addition, the cortices of Baloh’s mice contained about the same amount of poly-glycine-proline per total protein weight as did two FTD cases. Brown, in similar comparisons, found that poly-glycine-proline levels in his models matched the low end of the range detected in human cases. The researchers concluded that the animals make about the same amount of C9ORF72 products as people do.
The authors were hoping for phenotypes reminiscent of ALS or FTD, but had no such luck. The mice moved normally and socialized properly with other mice. Baloh’s proficiently navigated mazes as well. They lived normal lifespans. When the scientists examined their tissues, they saw healthy neuroanatomy, with no aggregates of disease-linked proteins such as TDP-43 or ubiquitin. “We did not see phenotypic abnormalities that would be convincing of neurodegeneration going on in young mice, or aged mice,” Baloh said.
“It gives you reason to pause about calling the foci or dipeptide repeats toxic,” commented Jeffrey Rothstein of Johns Hopkins University in Baltimore, even though in other model systems those poly-dipeptides clearly damage cells. However, Baloh said these mice do not prove that repeat RNAs and poly-dipeptides are wholly innocuous, just that these concentrations, in mice, do not cause neurodegeneration. He noted that it is common for mouse models to incompletely recapitulate human disease. He speculated the models might parallel a person at the presymptomatic stage of C9ORF72 disease. Mice, with their brief two-year lives, simply may not age long enough for the effects of C9ORF72 RNAs and poly-dipeptides to develop. “I am happy with the way they came out,” Baloh said of the animals. “I think they are an accurate model of the human levels of expression and burden of pathology.”
Baloh surmised that a second hit from a stressor such as defective proteostasis might instigate disease. His lab has begun experiments in cell culture to test that hypothesis. Both Baloh and Mueller also suggested that in people, the disease might result from a combination of the repeat RNAs or poly-dipeptides, plus the loss of function of the normal C9ORF72 gene. In that case, scientists would have to cross these gain-of-function models with C9ORF72 knockouts to see the full scope of the disease in mice.
Notably, other scientists have observed degeneration in their C9ORF72 model mice. Leonard Petrucelli (a co-author on both Neuron papers) used a virus to deliver the repeats and overexpress them in mice, yielding a neurodegenerative phenotype (see May 2015 news). Laura Ranum at the University of Florida in Gainesville has also generated mice with a full-length transgene, and they show signs of motor neuron disease as well.
What’s the difference? It will be interesting to compare the mice once they have been published, Baloh said. He and Mueller both speculate that expression level might determine phenotypes. Christopher Donnelly of the University of Pittsburgh (who has no mouse in the race) said he would have predicted that the full-length transgene was required to produce neurodegeneration. However, Baloh’s model negates that hypothesis.
Regardless of the mice’s apparent robust health, scientists agreed that the animals have value. “They are wonderful models of pathology … a great tool to test therapeutics,” Donnelly said. Each research group made initial forays in that direction using primary neural cultures from the animals. Baloh’s lab suppressed the RNA foci and poly-dipeptides with antisense oligonucleotides, while Brown’s did so with artificial microRNAs (see Nov 2015 conference news). Rothstein agreed the mice make good testing grounds for therapeutics, though he lamented the fact that they will not be useful to understand how C9ORF72 repeats cause neurodegeneration. Donnelly also noted that it will be useful to study these mouse lines because they have the human transgene and the endogenous mouse C9ORF72 as a backup. Scientists could try to edit out the repeats, or modulate expression of the human gene, without worrying that the mice would suffer from lack of normal C9ORF72 function.

Nucleolar exodus.
Nucleolin fills the entire nucleus in brain tissue from C9ORF72 model mice (bottom), compared to the nucleolus in controls (top). [Neuron, O’Rourke et al.]
Baloh and colleagues also assessed the status of the nucleolus in their model, noting that several studies point to nucleolar defects as a potential factor in C9ORF72-based disease (see Aug 2014 news; Dec 2014 news). They observed nucleolin, a protein involved in ribosome assembly in that organelle, redistributed across the entire nucleus in brain tissues from the model mice (see image at left). The same occurs in human tissues from C9ORF72 expansion carriers (see Mar 2014 news). However, they did not find any further defects, such as problems with ribosomal RNA production. More recently, scientists suggested that C9ORF72 expansion products interfere with nucleocytoplasmic transport, and Baloh will collaborate with Rothstein to examine transport in his model, he said (see Aug 2015 news).
“The two new C9 repeat models are intriguing and mark a significant advancement in ALS research,” commented Ronald Klein of the Louisiana State University Health Sciences Center in Shreveport, who was not involved in the study. “In the absence of an overt behavioral phenotype or neuronal loss, it will be interesting to see if the mice harboring the repeats are most susceptible to toxic treatments or overexpression of TDP-43.”—Amber Dance
References
News Citations
- C9ORF72 Mice Point to Gain of Toxic Function in ALS, FTD
- First C9ORF72 Mice Mimic Key Pathology, Behavior
- Listen Up, Gene Silencing Strikes a Chord at RNA Meeting
- C9ORF72 Killer Dipeptides Clog the Nucleolus
- Live-Cell Studies Blame Arginine Peptides for C9ORF72’s Crimes
- RNA-DNA Pairs: At the Root of C9ORF72 Repeat Damage?
- ALS Gene Repeats Obstruct Traffic Between Nucleus and Cytoplasm
Further Reading
Papers
- Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D, Neumann M. Quantitative analysis and clinico-pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol. 2015 Dec;130(6):845-61. Epub 2015 Sep 15 PubMed.
- Mackenzie IR. The role of dipeptide-repeat protein pathology in C9orf72 mutation cases. Neuropathol Appl Neurobiol. 2016 Apr;42(3):217-9. PubMed.
- Davidson Y, Robinson AC, Liu X, Wu D, Troakes C, Rollinson S, Masuda-Suzukake M, Suzuki G, Nonaka T, Shi J, Tian J, Hamdalla H, Ealing J, Richardson A, Jones M, Pickering-Brown S, Snowden JS, Hasegawa M, Mann DM. Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP-43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol Appl Neurobiol. 2016 Apr;42(3):242-54. Epub 2015 Dec 7 PubMed.
Primary Papers
- O'Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson DW, Harms MB, Petrucelli L, Lee EB, Lutz CM, Baloh RH. C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD. Neuron. 2015 Dec 2;88(5):892-901. PubMed.
- Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, Sun H, Boylan KB, Dickson D, Kennedy Z, Lin Z, Zhang YJ, Daughrity L, Jung C, Gao FB, Sapp PC, Horvitz HR, Bosco DA, Brown SP, de Jong P, Petrucelli L, Mueller C, Brown RH Jr. Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice. Neuron. 2015 Dec 2;88(5):902-9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.