For FTD Drug Development, a Q&A With Regulators
Quick Links
In advance of a 4 June meeting in Washington, D.C., with regulators and industry scientists, Adam Boxer of the University of California, San Francisco, and other steering committee members from the Frontotemporal Degeneration Study Group submitted questions to the U.S. Food and Drug Administration (FDA). FDA scientists including Russell Katz of the Center for Drug Evaluation and Research (CDER) and Nicholas Kozauer from the center’s Degeneration Group huddled over those questions. As they were unable to attend the meeting, Sampaio agreed to present their answers, adding her own view in places. FTD clinical trial pioneers, here’s the redux:
Q: Compounds have entered clinical trials for the treatment of progressive supranuclear palsy (PSP) predicated on the notion that PSP represents a fairly homogeneous tauopathy. What is your view regarding the generalization of clinical trials from PSP to other sporadic tauopathies such as corticobasal degeneration (CBD), non-fluent/agrammatic variant primary progressive aphasia (nfvPPA) and behavioral variant FTD (bvFTD)? Do you need randomized clinical trials in other tauopathies, or would you consider biomarker confirmation of a drug effect on tau?
A: Tauopathy as a unified indication across clinical disorders is problematic unless you have a single outcome measure for them. At least one separate study would likely be required for each disease. The relationship between pathology and clinical presentations in these diseases is still emerging. One study in a new indication may be sufficient if a widely accepted relationship with PSP exists. This all presupposes that a biomarker for tau exists.
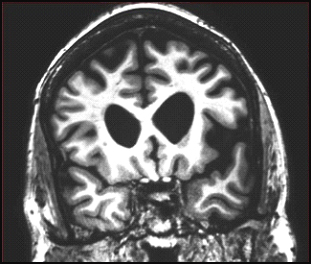
Coronal MRI scan through frontal lobes of a 62-year-old patient with the agrammatic variant of primary progressive aphasia demonstrating frontal lobe atrophy (on the left side of the brain, right side of the image). Image courtesy of Jason Warren
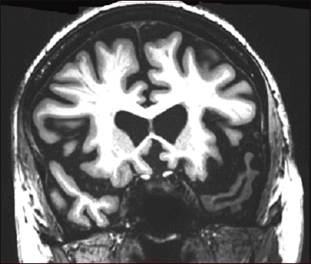
Coronal MRI scan through frontal lobes of a 66-year-old patient with the semantic variant of primary progressive aphasia demonstrating temporal lobe atrophy (on the left side of the brain, right side of the image). Image courtesy of Jason Warren
Q: Given the clinical and pathological heterogeneity of other sporadic tauopathies, would you recommend the use of enrichment strategies in trials, for example, a biomarker to identify pathology as a determinant for inclusion in the study?
A: Absolutely.
Q: Does the agency believe a non-specific label for the treatment of sporadic tauopathies is possible, analogous to neuropathic pain?
A: Neuropathic pain is a syndrome; its pathology is unknown. Ultimately, this is the surrogate question. You would have to demonstrate an understanding of the actions of the drug and the given disease.
Sampaio: The FDA does not give umbrella indications for brain syndromes; the EMA may be more likely to do that. It is too early to make the case that when we treat tau, we treat disease. Personally, I think that is where we are headed.
Q: Do you have a view regarding the current diagnostic criteria for the sporadic tauopathies?
A: They appear reasonable from a regulatory perspective.
Q: Do you agree that the diagnoses of PSP, FTD, and CBD are sufficiently well established to represent treatment indications for new therapies?
A: Yes.
Q: Regarding clinical trials in CBD, bvFTD, or nfvPPA, do you have a particular view on specific versus global outcome measures relevant to the diseases being studied? What about the need for co-primary endpoints?
A: Expect similar guiding principles as in symptomatic AD.
Sampaio: I personally take a different view. The CGI performs poorly in long trials. First of all, "CGI" stands for clinical global impression. In a long-term trial of a neurodegenerative disease, you do not expect improvement, so even the title of the scale is wrong. Secondly, the investigator is anchored to the baseline that can be 18 months earlier, so this, in effect, becomes a memory test for the rater. I think the FTSG should define innovative designs and create the data to support your proposals. You are the experts. Bring it to the agency; don't fight among yourselves, and then persuade us with a good argument.
Q: Do you have a view on what the clinically relevant benefit would be in these conditions?
A: No. That is why we require a global or functional co-primary endpoint.
Q: Do you have a view on the degree to which tau-directed therapies need to affect some or all isoforms, 3R versus 4R?
A: No. We are concerned with clinical outcome versus specified mechanism of action. We cannot comment on the acceptability of an effect on a given tau isoform as a surrogate at this point.
Q: Regarding translation of findings from PSP studies to Alzheimer’s patients, would positive PSP studies preclude the need for dose ranging in AD trials?
A: No. A relationship cannot be assumed.
Q: Would positive PSP studies provide supportive safety data for AD studies and reduce the need for long-term exposure in AD patients?
A: Yes.
Q: Would your view regarding the value of CSF tau/p-tau as a validated biomarker in AD change if treatment for PSP affected CSF tau/p-tau?
A: No. Again, the relationship cannot be assumed.
Q: With regard to developing a treatment for FTD in patients with progranulin mutations, how large a sample is needed to meet regulatory needs for safety?
A: There is no pre-specified exposure requirement for safety data in orphan conditions. We can be flexible.
Q: What outcomes might be convincing in presymptomatic carriers? Would raising serum progranulin suffice for conditional approval? If not, would a drug-placebo difference on a single composite measure relevant to disease be sufficient?
A: This is, again, the surrogate question. It requires a very detailed understanding of the MOA of both drug and disease. What would be the composite measure in presymptomatic disease? We are open to considering an argument, as in early stages of AD, where the impetus is toward specific cognitive outcomes, not global clinical ones.
Sampaio: This, in my view, is the most important trial. We have discussed this extensively in AD [for regulatory coverage, see ARF DIAN story, ARF related news story, ARF API story].
Q: Preliminary work exists on clinical trial measures for FTD or tau populations. Some tools, such as the NPI, have been used extensively in AD trials as secondary outcomes. The CDER has been expanded to include domains relevant to FTD for use in FTD trials. How much validation would be required to accept those as an outcome measure for a clinical trial?
A: Face validity may be reasonable in otherwise accepted scales.
All told, the regulatory experts sent the FTSG on its way with these closing comments: “We want products for sufferers of rare diseases that actually work, so we want to base our decision on the best science. We are excited to work with any and all of you to figure out the most efficient path. Fast is not always most efficient,” said Yao. Sampaio concurred. She closed the day with a last point that industry had brought up in side conversations, as well. “People get excited about biomarkers. But remember that biomarkers are not interchangeable until you prove the relationships between them,” Sampaio said. “I see speakers make lists of imaging modalities and fluid outcomes, and then assign group sizes to each. Doing that is a recipe for failure. This is not a supermarket where you can shop the one that gives you fewer patients in the trial. Each marker measures a different thing, and you must understand how they relate.”—Gabrielle Strobel.
This concludes a five-part series. See Part 1, Part 2, Part 3, Part 4. Read a PDF of the entire series.
References
News Citations
- DIAN: What Sayeth the Regulator? Q&A With Rusty Katz
- London: What Regulators Say About Trials in Familial AD
- Scientists and Regulators Discuss Preclinical AD Trials
- Scientists Strategize With Regulators for Frontal Assault on FTD
- Toward FTD Therapeutic Trials: Diagnosis Firm, Outcomes Still Soft
- Case Studies Crystallize Trial Ideas at FTD Conference
- Take It From the Agency: First FTD Drug May Be an Orphan
Other Citations
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.