Exosomes: Purveyors of Neurodegenerative Disease?
Quick Links
See Part 5 and Part 7 of this series.
Exosomes have emerged as players in cell-to-cell communication in the nervous system. At the Society for Neuroscience annual meeting held in Washington, D.C., November 15 to 19, scientists reported that these tiny, cell-derived vesicles may also contribute to pathology in neurodegenerative disease. Exosomes could make Aβ and carry it, and other pathogenic proteins such as tau, from one neuron to another, the story goes. At the same time, blocking the release of exosomes may lead to an accumulation of toxic proteins inside neurons, contributing to pathology.
Aβ-Generating Machines?
Exosomes are tiny packets of proteins, lipids, and nucleic acids that are released from most cells in the body. They form when the endosomal membrane folds in on itself and pinches off to create a vesicle inside the endosome’s lumen. Dozens of them jostle in endosome-derived multivesicular bodies. When those bodies fuse with the plasma membrane, the vesicles flood the extracellular space as exosomes (for details, see Part 5 of this series).
At SfN, Lawrence Rajendran, University of Zurich, explained that exosomes in neurons convene all of the working parts needed to make the Aβ peptide. He previously found that exosomes from neuroblastoma cells contained the β C-terminal fragment (CTF) of the amyloid precursor protein (APP) and Aβ. Others had reported that exosomes also contained β-secretase, but whether it was active was unclear (see Sharples et al., 2008). Could β- and γ-secretases possibly produce Aβ outside the cell? Studying exosome fractions from mouse primary neurons, Rajendran and colleagues have so far found BACE1 and all four components of the γ-secretase complex.
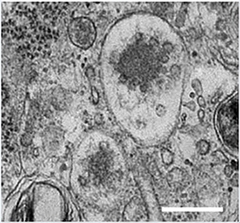
Exosomes and Parkinson's.
Exosomes accumulate as small vesicles in multivesicular bodies. Genetic variants in the PARK9 gene that cause an early form of Parkinson's limit their production (see text below). [Image courtesy of Tsunemi et al., The Journal of Neuroscience, 2014.]
Though preliminary, their evidence suggests that the γ-secretase is active, because when incubated with APP β-CTF, the exosomes produced Aβ. No Aβ formed in the presence of a γ-secretase inhibitor, or when the exosomes came from mice lacking presenilins. This hints that exosomes carry the machinery for making Aβ. “We don’t know whether this is physiologically relevant, but it is an interesting observation,” Rajendran said. He emphasized that there is no evidence that exosomes make Aβ outside the cell in vivo. However, since amyloid-forming proteins are more prone to aggregation when they associate with membranes (see Butterfield and Lashuel, 2010), any exosome-associated Aβ could seed the aggregation of the peptide outside the cell, he said.
Do Exosomes ‘Pass It On’?
If exosomes offer cells a way to get rid of Aβ, then they could spread the peptide throughout the brain. For now, there is no evidence that neurons take up vesicles with pathogenic proteins in them, said Rajendran. Martin Hallbeck, Linköping University, Sweden, is studying this question in neurons derived from induced pluripotent stem cells made from healthy human fibroblasts. He incubated these neuron cultures with exosomes from neuroblastoma cells laden with fluorescently labeled α-synuclein or Aβ oligomers. Using confocal microscopy, he saw neurons ingest the exosomes and their proteins. When he then co-cultured these cells with fresh neurons that had not been incubated with exosomes, he saw that labeled oligomers of both α-synuclein and Aβ passed between them via exosomes. However, if he blocked endocytosis, the cells took up almost none. “These results suggest that exosomes provide at least one way for toxic aggregates to spread from neuron to neuron,” Hallbeck told Alzforum. While he and his research group had previously shown that oligomers transferred directly between neurons were toxic, he did not examine whether the exosome-delivered oligomers were, as well.
Neurons may not be the only ones propagating neurodegenerative proteins via exosomes. Tsuneya Ikezu, Boston University School of Medicine, believes microglia may do it, too. He thinks that microglia take up pathogenic aggregates from outside the cell and then spit some back out in exosomes, perhaps because they are hard to break down. To test this idea, Ikezu’s team injected the medial entorhinal cortex of wild-type mice with a virus encoding human tau. A special reporter that labeled virus-infected cells green distinguished them from the cells that might take up just transgenic tau. The human tau spread to the dentate gyrus over 28 days. Before it accumulated there, however, Ikezu noted activated microglia in the region, suggesting to him that neurons were sending advance warning along axons that terminate in the dentate gyrus, recruiting microglia. He thinks microglia took up the human tau and released what they were unable to digest in exosomes to be passed to other cells. In this experiment, preventing microglial activation greatly reduced tau spread in the mice. So did inhibiting neutral sphingomyelinase-2, which is required to make exosomes. Immunoelectron microscopy also revealed human tau inside the exosomes from the mouse brain.
This talk received mixed reviews. Dave Morgan, University of South Florida, Tampa, found it fascinating that reducing microglial activation slowed tau propagation. “Of course, there are many other reasons why microglial depletion could reduce tau spread, but this [elimination of microglial exosomes] is looking like a potential candidate mechanism,” he told Alzforum. Greg Cole, University of California, Los Angeles, also thought the findings were interesting. “This is a very new idea and the data to support it are novel and intriguing,” he wrote. However, he wondered if inhibiting neutral sphingomyelinase-2 could have similar effects on tau spread between neurons in the absence of a microglial effect. Cole noted that reducing microglial activation could reduce tauopathy independently of exosomes, for example, by diminishing tau phosphorylation.
Too Many Exosomes Are Bad, Too Few Even Worse
Rocío Pérez-González and Sebastien A. Gauthier, working in the lab of Efrat Levy at the Nathan S. Kline Institute, Orangeburg, New York, showed that brain cells from the postmortem tissue of people with Down’s syndrome expelled more exosomes than cells from people who had been healthy. Each vesicle also contained more APP and APP-CTFs. The researchers reported that cells in the Ts2 mouse model of Down’s do the same. Pérez-González suspects that a backup in the endosomal pathway forces the cell to release exosomes as a protective mechanism. “If the exosomes are enriched with toxic material, they may also be pathogenic and could help spread disease all over the brain,” she told Alzforum. The finding is in line with other research suggesting that diseased cells produce more exosomes, said Francesca Properzi, Superior Institute for Health, Rome. While the exosomes could result from a backed-up endosomal system, they could also simply serve as a way for cell to rid themselves of toxins.
Secreting too few exosomes could also pose a problem. Researchers led by Dimitri Krainc, Northwestern University, Chicago, have reported that α-synuclein accumulates inside fibroblasts from people with Kufor–Rakeb syndrome, an early onset form of Parkinson’s disease. Their fibroblasts make fewer intraluminal vesicles and release fewer exosomes than do cells from controls. Kufor-Rakeb results from a mutation in the PARK9 gene, a member of a family of ATPases that transport ions across membranes. Krainc found that overexpressing the PARK9 protein in mouse primary cortical neurons had the opposite effect, causing the cells to release synuclein into the medium. The researchers think PARK9 interacts with proteins of the endosomal sorting complex, which is required for making exosomes. They published this work in the November 12 Journal of Neuroscience (see Tsunemi et al., 2014). PARK9 defects were previously linked to lysosomal dysfunction (see Jun 2012 news story on Dehay et al., 2012).
Given that exosomes appear to be mixed up in neurodegenerative disease, might they offer a therapeutic target? Since exosomes appear to rid cells of waste, interfering with their entry into recipient cells might be better than inhibiting their release. Xandra Breakefield, Massachusetts General Hospital, Charlestown, who co-chaired the session with Rajendran, said that if specific interactions mediate exosome uptake into particular cells, those interactions could be selectively blocked. Very little is known about uptake mechanism at this point. Some groups are working to develop exosomes as biomarkers of neurodegenerative diseases, including AD and PD (see Aug 2014 news story; and Shi et al., 2014), recommended that more basic biology be studied before trying to target these vesicles.—Gwyneth Dickey Zakaib
References
News Citations
- Exosomes: The FedEx of the Nervous System?
- Hats Off to ApoE—Key to Formation of Functional Amyloid
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- Exosomes Stand Out as Potential Blood Biomarkers
Paper Citations
- Sharples RA, Vella LJ, Nisbet RM, Naylor R, Perez K, Barnham KJ, Masters CL, Hill AF. Inhibition of gamma-secretase causes increased secretion of amyloid precursor protein C-terminal fragments in association with exosomes. FASEB J. 2008 May;22(5):1469-78. PubMed.
- Butterfield SM, Lashuel HA. Amyloidogenic protein-membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed Engl. 2010 Aug 2;49(33):5628-54. PubMed.
- Villar AJ, Belichenko PV, Gillespie AM, Kozy HM, Mobley WC, Epstein CJ. Identification and characterization of a new Down syndrome model, Ts[Rb(12.1716)]2Cje, resulting from a spontaneous Robertsonian fusion between T(171)65Dn and mouse chromosome 12. Mamm Genome. 2005 Feb;16(2):79-90. PubMed.
- Tsunemi T, Hamada K, Krainc D. ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci. 2014 Nov 12;34(46):15281-7. PubMed.
- Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, Vital A, Vila M, Klein C, Bezard E. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012 Jun 12;109(24):9611-6. PubMed.
- Shi M, Liu C, Cook TJ, Bullock KM, Zhao Y, Ginghina C, Li Y, Aro P, Dator R, He C, Hipp MJ, Zabetian CP, Peskind ER, Hu SC, Quinn JF, Galasko DR, Banks WA, Zhang J. Plasma exosomal α-synuclein is likely CNS-derived and increased in Parkinson's disease. Acta Neuropathol. 2014 Nov;128(5):639-50. Epub 2014 Jul 6 PubMed.
Further Reading
News
- Evidence Piles Up for Lysosomal Dysfunction in Parkinson’s
- The Epidemic in Your Head? New Model Casts Amyloid as Intra-Brain Contagion
- Huntington’s Protein Charged with Trans-synaptic Spread of Pathology
- Tau, α-Synuclein Spread: Crazy Stuff—How Might It Work?
- Mice Tell Tale of Tau Transmission, Alzheimer’s Progression
- In Case You Wondered: Neurodegenerative Diseases Are Not Contagious
Primary Papers
- Tsunemi T, Hamada K, Krainc D. ATP13A2/PARK9 regulates secretion of exosomes and α-synuclein. J Neurosci. 2014 Nov 12;34(46):15281-7. PubMed.
- Rajendran L, Bali J, Barr MM, Court FA, Krämer-Albers EM, Picou F, Raposo G, van der Vos KE, van Niel G, Wang J, Breakefield XO. Emerging roles of extracellular vesicles in the nervous system. J Neurosci. 2014 Nov 12;34(46):15482-9. PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.