Cytokine Takes Aβ Off the Menu for Microglia
Quick Links
Microglia may like to dine on amyloid plaques, but does the immunosuppressive environment in the brain quash their appetite? Apparently so. According to two recent studies, lifting the suppression unleashes the microglia’s craving, allowing them to feast on plaques, but without sampling neurons. The studies—one led by Todd Golde at the University of Florida in Gainesville and the other by Terrence Town at the University of Southern California in Los Angeles—used complementary approaches to reveal that the anti-inflammatory cytokine IL-10 worsens Alzheimer’s disease pathology and cognitive decline in mouse models. Removing the cytokine slowed disease pathology and kept synapses intact. Kevin Doty presented findings from the Town lab at “Neuroinflammation in Diseases of the Central Nervous System,” a Keystone symposium held January 25-30 in Taos, New Mexico. That work appeared in the February 4 Neuron, while Golde's was published in the January 7 issue of the same journal.
“It turns out that you can have your cake and eat it too,” Town told Alzforum. “Revving up microglia removes plaques without damaging neurons.”
Both labs had already shared preliminary results from their studies at conferences covered by Alzforum (see Mar 2013 and Dec 2013 conference coverage). In their published form, the studies also revealed that IL-10 boosts astroglial expression of ApoE, which the researchers reported decreases microglial appetite for Aβ in vitro. Other data presented at Keystone put ApoE as a major factor in promoting a pro-inflammatory microglial phenotype (see Part 3 of this series).
Neuroinflammation plays an important role in clearing the brain of debris, infectious pathogens, and sickly cells. However, if uncontrolled or chronic, it can also harm the brain, and this can occur during neurodegenerative disease (see Wyss-Coray and Mucke, 2002; Perry, 2010).
Neuroinflammation manifests as a complex yin-yang relationship between anti- and pro-inflammatory responses, and much previous work has focused on the latter. Some have reported that shutting down pro-inflammatory cytokines lessens AD symptoms (see Kiyota et al., 2010; Jun 2008 news; and Nov 2012 news). Other labs, including Golde’s, have reported that overexpressing pro-inflammatory cytokines leads to plaque clearance (see Boissonneault et al., 2009; Chakrabarty et al., 2010; Chakrabarty et al., 2010; Chakrabarty et al., 2011).
The quieter role of the yin—the anti-inflammatory cytokines—had been less explored. Two major anti-inflammatory cytokines, TGF-β and IL-10, are known to shut down immune responses before they spin out of control. In 2008, Town generated AD mice lacking TGF-β activity in macrophages and microglia. He reported that mice lacking this wet-blanket cytokine had fewer plaques and less cognitive decline than mice with TGF-β. Importantly, this occurred without damage to neurons (see Jun 2008 news). The result encouraged Town to go after IL-10.
In Town’s study, first author Marie-Victoire Guillot-Sestier and colleagues crossed IL-10-deficient mice with APP/PS1 mice. They found that year-old APP/PS1 mice harbored far fewer plaques when they lacked the cytokine. In biochemical analysis, soluble and insoluble Aβ40 and Aβ42 plummeted by more than half. In brain slices, activated microglia crowded around any remaining plaques, which appeared more diffuse than in animals with the cytokine. These cells expressed low levels of CD45, suggesting that they were resident microglia rather than infiltrating macrophages from outside the brain (see Part 2, Part 3, and Part 4 for more on the nature of immune cells in the AD brain). Immunostaining also revealed that these cells contained Aβ within phagolysosomes. When put into a dish, microglia harvested from IL-10-deficient mice more readily gobbled up aggregated Aβ42 than normal microglia did, and they sported more markers of activation.
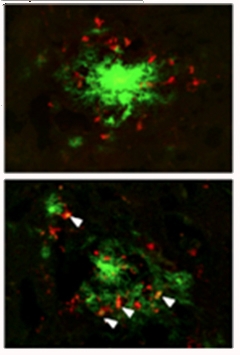
Feeding Frenzy?
Activated microglia (red) from APP/PS1 mice lacking IL-10 (bottom) more readily clustered around plaques (green) than microglia from control APP/PS1 mice (top). [Image courtesy of Guillot-Sestier et al., Neuron, 2015.]
Importantly, despite their penchant for destroying plaques, microglia in IL-10-deficient mice left neurons alone. In fact, these mice had wild-type levels of the synaptic marker synaptophysin, suggesting that synapses were preserved. This paid off in some cognitive benefits. The mice more readily recognized novel objects (a signature of episodic memory) than APP/PS1 mice, and they were less hyperactive. However, IL-10 deficiency did not rescue APP/PS1 mice from deficits in spatial working memory.
To take stock of the changes wrought in the IL-10 deficient mice, the researchers compared their gene expression with that of APP/PS1 mice. They identified 117 differentially expressed genes, the majority involved in innate immune function, chemoattraction, Aβ interaction, or phagocytosis. Surprisingly, the researchers found that IL-10-deficient APP/PS1 mice made less ApoE.
Could a dearth of ApoE explain the microglia's taste for Aβ? ApoE influences clearance of different forms of Aβ, but it is not clear how (see Apr 2013 news). The researchers tested whether ApoE would affect microglial uptake of Aβ. ApoE2 barely altered uptake of Aβ aggregates, whereas ApoE3 and ApoE4 reduced it by about 70 percent. Importantly, IL-10-deficient microglia efficiently consumed Aβ when mixed with either ApoE2 or ApoE3, but not ApoE4 (see Alzforum Webinar).
In Taos, Gary Landreth of Case Western Reserve University in Chicago noted that it was unclear how ApoE was inhibiting Aβ uptake in this tissue culture model, and he questioned whether the experiments were modeling bona fide phagocytosis.
Golde’s lab took the reciprocal approach. Rather than knocking out IL-10, first author Paramita Chakrabarty and colleagues overexpressed the cytokine in the mouse brain. The researchers used a method they had previously pioneered to overexpress other cytokines. They injected adeno-associated virus 2/1 (AAV2/1) expressing IL-10 directly into the brain of neonatal CRND8 mice.
For the most part, the results of Golde’s study mirrored Town’s. Overexpression of IL-10 elevated plaque burden, damaged synapses, and accelerated cognitive decline. Microglia huddled around plaques but cleared little Aβ. A comparison of whole-brain gene-expression profiles revealed 140 genes differentially expressed between CRND8 mice with or without IL-10 overexpression, including chemokines, complement, and immune signaling genes. ApoE was upregulated 1.7-fold in CRND8 mice overexpressing IL-10.
As had Town’s group, Chakrabarty and colleagues decided to investigate the link between IL-10 and ApoE. The researchers found that ApoE protein levels rose nearly fourfold in the insoluble fraction of brain extracts of mice overexpressing IL-10, and immunofluorescence showed that ApoE was expressed by astrocytes and intermixed with plaques. To measure ApoE’s effects on Aβ uptake by microglia, the researchers incubated the cells in media taken from IL-10-deficient astrocyte cultures, which are chock-full of ApoE. When they fed the cells aggregated, fluorescent Aβ, they saw a reduction in microglial uptake compared to cells grown in normal astrocyte media.
Town said it was exceedingly rare that two papers agreed to this extent. “It’s really uncanny,” he said. Both Town and Golde propose a mechanism whereby microglia secrete IL-10, which then prevents them from taking up Aβ. In parallel, IL-10 boosts ApoE expression, which could reduce Aβ uptake even more.
Could blocking IL-10 work as a therapy for AD? Milan Fiala of the University of California, Los Angeles, found the results intriguing, but commented that blocking IL-10 in people would likely have inconsistent effects, as background levels of inflammatory and anti-inflammatory cytokines vary markedly throughout the population.
Richard Ransohoff of Biogen Idec in Cambridge, Massachusetts, agreed with Fiala, noting that blocking IL-10 could trigger harmful inflammatory effects. He added that results based on the manipulation of a single cytokine, such as IL-10, could be too simplistic to support therapeutic conclusions.—Jessica Shugart
References
News Citations
- Blessing or Curse? Peripheral Cytokines in the Brain
- Innate Immune Cells Enlisted to Clear Amyloid, Fight Disease
- Microglia in Disease: Innocent Bystanders, or Agents of Destruction?
- Complement: AD Friend or Foe? New Work Tips Balance to Former
- Soothing Neuroinflammation Quells Plaques in Mice
- Macrophages Storm Blood-brain Barrier, Clear Plaques—or Do They?
- Nature Versus Nurture: What Gives Microglia Their Identity?
- United in Confusion: TREM2 Puzzles Researchers in Taos
- ApoE Does Not Bind Aβ, Competes for Clearance
Research Models Citations
Webinar Citations
Paper Citations
- Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 2002 Aug 1;35(3):419-32. PubMed.
- Perry VH. Contribution of systemic inflammation to chronic neurodegeneration. Acta Neuropathol. 2010 Sep;120(3):277-86. PubMed.
- Kiyota T, Okuyama S, Swan RJ, Jacobsen MT, Gendelman HE, Ikezu T. CNS expression of anti-inflammatory cytokine interleukin-4 attenuates Alzheimer's disease-like pathogenesis in APP+PS1 bigenic mice. FASEB J. 2010 Aug;24(8):3093-102. PubMed.
- Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer's disease. Brain. 2009 Apr;132(Pt 4):1078-92. Epub 2009 Jan 17 PubMed.
- Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, Zubair AC, Dickson D, Golde TE, Das P. Massive gliosis induced by interleukin-6 suppresses Abeta deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010 Feb;24(2):548-59. PubMed.
- Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, Dickson D, Golde TE, Das P. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010 May 1;184(9):5333-43. PubMed.
- Chakrabarty P, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol Neurodegener. 2011;6:16. PubMed.
Further Reading
Primary Papers
- Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, DiNunno N, Rosario AM, Cruz PE, Verbeeck C, Sacino A, Nix S, Janus C, Price ND, Das P, Golde TE. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015 Feb 4;85(3):519-33. Epub 2015 Jan 22 PubMed.
- Guillot-Sestier MV, Doty KR, Gate D, Rodriguez J Jr, Leung BP, Rezai-Zadeh K, Town T. Il10 deficiency rebalances innate immunity to mitigate Alzheimer-like pathology. Neuron. 2015 Feb 4;85(3):534-48. Epub 2015 Jan 22 PubMed.
Annotate
To make an annotation you must Login or Register.
Comments
Indiana University School of Medicine
There is considerable confusion over the roles of neuroinflammation in animal models of Alzheimer’s disease and other neurodegenerative disorders. The complementary studies of Chakrabarty et al. and Guillot-Sestier et al. have shed new light, but not necessarily enhanced understanding, of the roles of IL-10 in AD pathogenesis. The anti-inflammatory cytokine IL-10 was generally thought to have salutary effects in mouse models of AD, based on a modest literature. The two new studies have provided compelling evidence that IL-10 acts to exacerbate amyloid pathology and impair behavior. Thus, we have fundamentally misunderstood the in vivo actions of this cytokine. Clearly, this is not the only immune effector whose actions in the brain have not conformed to their established roles in the periphery. These compelling and provocative studies serve to highlight the turmoil in understanding the complex actions of immunity in the normal and diseased brain. The field as a whole is undergoing a resurgence in interest owing to new genetic linkages of immune genes to various neurodegenerative diseases. However, as the current work highlights, loss- and gain-of-function interventions have yielded unpredicted and not readily interpretable outcomes. And our notions of pro- and anti-inflammatory actions, based largely on studies in peripheral macrophages, have proven to be false.
Both papers report IL-10 regulation of ApoE expression in the brain, which was not previously appreciated. Similarly, they postulate that phagocytosis is suppressed by IL-10. The linkage between these two observations was explored by Guillot-Sestier et al. who found IL-10- and ApoE-isoform-dependent microglial phagocytosis, and the basis of this latter effect is unexplained. Chakrabarty et al. report an ApoE-stimulated uptake of Aβ (and it is unclear whether this was murine or human ApoE), using an unusual experimental design. While the new data strongly suggests that ApoE may participate in Aβ uptake by microglia, I think the jury is still out, and this point requires additional investigation.
UCLA
This is nice intricate work in a model system on the importance of inflammation in clearing amyloid-β (Aβ), as shown indirectly through improvement of clearance by blockade of IL-10. The role of inflammatory activation of macrophages/microglia and phagocytosis of Aβ has been investigated in patients with AD and controls for years (Fiala et al., 2001, 2002, 2007, 2009, 2015). The effect of the blockade of IL-10 is another piece of evidence that inflammation can be both detrimental and beneficial. However, inflammation in animal models has low correlation with human inflammation (Seok et al., 2013). The patients with AD and mild cognitive impairment are very heterogeneous, either “inflammatory” with a high level of inflammation, or “non-inflammatory” with a low level of inflammation. Any single approach is not going to fit all, and modulation of cytokines over long periods has unknown consequences. It is clear from our studies that phagocytosis of Aβ is defective in all AD and MCI patients (unless supplemented with omega-3); whereas it is excellent in some cognitively normal persons into very old age. Thus physiological mechanisms exist which balance inflammation and phagocytosis. Omega-3 fatty acid-derived specialized proresolving mediators (resolvins, protectins and maresins) both terminate inflammation and perform resolution (Serhan and Petasis, 2011). Extensive studies of omega-3 fatty acids are ongoing in various disorders. Our recent study shows that omega-3 fatty acid supplementation in MCI patients with a daily drink, Smartfish, increases phagocytosis of Aβ and resolvin D1 in macrophages (Fiala et al., 2015). Omega-3 supplementation is a practical approach for improving amyloid-beta immunity in MCI patients, which seems to also stabilize the cognition (Freund-Levi et al., 2006).
References:
Fiala M, Liu QN, Reddy S, Graves MC. Macrophages infiltrate the brain and express COX-2 and iNOS in Alzheimer's disease and AIDS. Alzheimer's Rep. 2001;4(1):1-7.
Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, Vinters HV. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer's disease brain and damage the blood-brain barrier. Eur J Clin Invest. 2002 May;32(5):360-71. PubMed.
Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM, Sayre J, Zhang L, Zaghi J, Dejbakhsh S, Chiang B, Hui J, Mahanian M, Baghaee A, Hong P, Cashman J. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer's disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci U S A. 2007 Jul 31;104(31):12849-54. PubMed.
Avagyan H, Goldenson B, Tse E, Masoumi A, Porter V, Wiedau-Pazos M, Sayre J, Ong R, Mahanian M, Koo P, Bae S, Micic M, Liu PT, Rosenthal MJ, Fiala M. Immune blood biomarkers of Alzheimer disease patients. J Neuroimmunol. 2009 May 29;210(1-2):67-72. PubMed.
Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A. 2013 Feb 26;110(9):3507-12. Epub 2013 Feb 11 PubMed.
Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. 2011 Oct 12;111(10):5922-43. Epub 2011 Jul 18 PubMed.
Fiala M, Halder RC, Sagong B, Ross O, Sayre J, Porter V, Bredesen DE. ω-3 Supplementation increases amyloid-β phagocytosis and resolvin D1 in patients with minor cognitive impairment. FASEB J. 2015 Jul;29(7):2681-9. Epub 2015 Mar 24 PubMed.
Freund-Levi Y, Eriksdotter-Jönhagen M, Cederholm T, Basun H, Faxén-Irving G, Garlind A, Vedin I, Vessby B, Wahlund LO, Palmblad J. Omega-3 fatty acid treatment in 174 patients with mild to moderate Alzheimer disease: OmegAD study: a randomized double-blind trial. Arch Neurol. 2006 Oct;63(10):1402-8. PubMed.
Make a Comment
To make a comment you must login or register.