Can Induced Neurons Identify Early Signs of Neurodegeneration?
Quick Links
Researchers have linked dozens of genetic loci to late-onset Alzheimer’s disease and found dozens more that cause familial forms of AD and other neurodegenerative diseases. Teasing out how each contributes to pathology has proven a little more difficult. The effects of one single nucleotide are easily drowned out by the contributions of its 3 billion neighbors in the human genome. Enter induced pluripotent stem cells and CRISPR gene editing. Scientists are now using these technologies to make isogenic cells lines whose genomes often differ by just that one single-nucleotide polymorphism. At this year’s Alzheimer’s Association International Conference, held July 14–18 in Los Angeles, researchers showed how they used isogenic cell lines to uncover deficits in GABAergic signaling and lysosomal function caused by mutations in tau, and to identify new endosomal pathways taken by the cell-sorting protein SORLA, a key player in recycling of the amyloid precursor protein.
- Tau variants raise lysosome pH, limit their proteolysis.
- R406W tau also retunes GABAergic networks.
- SORLA sorts APP through non-retromer pathways.
Trouble with Tau
In LA, Celeste Karch, Washington University, St. Louis, described how she used isogenic cell lines to study tau mutations. Karch’s overarching goal is to explore how genetic variants that cause familial AD and frontotemporal dementia affect neuronal cell biology. Karch focused on the R406W mutation that causes autosomal dominant FTD. This variant of the disease carries the pathological hallmarks of a typical tauopathy, namely tangles in neurons and glia, but clinically manifests with progressive memory loss much like Alzheimer’s disease. Karch wants to identify molecular events downstream of the mutation that might be responsible.
Collaborating with Carlos Cruchaga and Oscar Harari, both also at WashU, Karch’s group started with skin fibroblasts donated by a R406W tau carrier who receives care at the Knight Alzheimer’s Disease Research Center. The researchers de-differentiated her fibroblasts into induced pluripotent stem cells, then used CRISPR to correct the tau gene back to wild-type in some of those cells. Next they converted the wild-type and R406W iPSCs into cortical neurons and used RNA-Seq to look for transcriptional differences (Jiang et al., 2018).
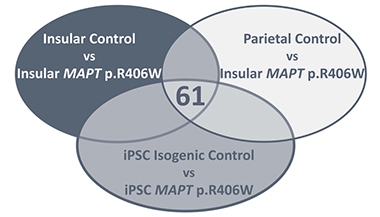
Common Changes. Comparing patient and control tissue against mutant and wild-type isogenic cell lines, researchers found common changes in expression of 61 genes. [Courtesy of Jiang et al., Translational Psychiatry 2018.]
The scientists found that expression of 328 genes differed between wild-type and R406W-tau-induced neurons. Which of those explain neurodegeneration in an older adult with FTD? Since iPSCs have been genetically “deprogrammed” back to an early embryonic state, some expression changes in neurons derived from iPSCs might reflect developmental rather than neurodegenerative states.
To address this problem, Karch’s team combined differential expression analysis of the iPSC-derived neurons with similar analyses of human tissue from the insular and parietal cortices of healthy controls and from people with the R406W mutation. This process identified 61 genes that are differentially expressed in both iPSCs and disease brain (see image above). Because the cell lines are isogenic, these expression differences are likely driven directly by the tau mutation.
What do the 61 do? Functional annotation analysis predicted that half of this cadre interacts with tau. Pathway analysis highlighted many genes related to calcium-dependent synaptic function and GABAergic signaling. The GABA-associated genes SNAP-25 and SYT1 emerged as hubs of an interaction network (see image below).
“Our data indicated that even though our [iPSC-derived] cells may still be ‘young,’ essentially patterning like developing fetal cells, tau mutations are sufficient to induce changes that are detectable in an older brain,” said Karch.

Dysfunctional Network? The R406W mutation in tau alters expression of a network of genes linked to calcium signaling and GABAergic transmission. [Courtesy of Jiang et al., Translational Psychiatry 2018.]
Knowing this, the researchers wondered what other changes the R406W variant might evoke. Sidhartha Mahali, a postdoc in Karch’s lab, noticed a curious phenotype when he grew iPSC-derived neurons in two-dimensional cell cultures. LAMP1-positive vesicles, aka lysosomes, spread much farther from the soma of R406W-tau neurons than they did from wild-type soma. This distribution might change how lysosomes function, said Karch. Other groups had shown that the farther lysosomes are from the nucleus, the weaker their capacity for degrading protein (Yap et al., 2018). Indeed, Mahali found that lysosomes in R406W-tau neurons were less acidic. They ramped up expression of proteolytic enzymes, yet had no appetite for degrading protein. The uptick in proteases seems to be an attempt to compensate for poor proteolysis, said Karch.
And how does tau fit in? In LA, Mahali reported that in R406W-tau neurons, both total tau and p231-tau accumulated in LAMP1-positive lysosomes. He was also able to recapitulate the lysosomal distribution phenotype and the tau co-localization when he generated neurons from iPSCs containing either the P301L or IVS10+16 tau mutations. All told, the data suggest that tau mutations somehow lead to a redistribution of lysosomes in the cell that may cause them to malfunction.
The talk sparked a lively discussion. Some wanted to know whether degradation of tau itself was affected by the lysosomal shift, and if there were any functional changes to the neurons. Karch has no data yet. She plans to use SILK isotope labelling to quantify tau production and clearance rates, and also plans to test if the mutations and the lysosome dysregulation affect neuronal excitability.
Others asked whether tau accumulates in lysosomes because the organelles clear too little of it, or if lysosomes malfunction because of the tau mutation. “That’s a great question,” said Karch. “We know tau gets degraded through several mechanisms in the cell, including through autophagy. However, whether mutant tau triggers degradative dysfunction remains uncertain.”
Joanna Jankowsky, Baylor College of Medicine, Houston, speculated that mutations in the microtubule binding protein disrupt transport of lysosomes. Karch said she was unsure about that. “It could be a transport issue or maybe a component of some signaling pathway is missing or malfunctioning. We are trying to parse that out,” she said.
Beth Stutzmann, Rosalind Franklin University of Medicine and Science, North Chicago, Illinois, suspects signaling. “We see calcium dysregulation upstream,” she told Alzforum. “When we restore Ca2+ levels we clear p-tau accumulation and return phagosome levels and lysosome function to normal.” Stutzmann, with first author Sarah Mustaly and others, reported that calcium dysregulation tones down lysosomal acidity, which in turn leads to accumulation of protein aggregates, including tau. They studied induced neurons derived from AD patient fibroblasts and in a mouse model of tau tangle and amyloidosis.
For the latter, Mustaly examined the hippocampi and cortices of 3-month-old 3xTG mice which express human APP and tau transgenes. She found reduced expression of the vacuolar ATPases that pump protons into the lysosome to keep their pH acidic. ATPase subunit loss came with more numerous mature autophagosomes, a sign lysosomes are not processing them properly. Levels of p262-tau also rose. Injecting the mice with the ryanodine receptor (RyR) modulator Ryanodex once daily for 30 days corrected not only levels of Ca2+, but also of V-ATPase, autophagosomes, and p-tau. RyRs sit in the endoplasmic reticulum membrane and pump Ca2+ into the cytosol. “We think that as the V-ATPases pumps protons into the lysosome, Ca2+ must be pumped out to maintain electrical balance,” said Stutzmann. “If the levels of Ca2+ in the cytosol increase, then the lysosome has to pump Ca2+ against a stronger gradient, and if they can’t then they become more alkaline,” she said. In keeping with this, Mustaly showed that a RyR agonist raised the lysosomes pH; this was countered by Ryanodex.
Might something similar be going on in the human brain? Mustaly generated induced neurons from fibroblasts donated by AD patients and healthy controls, and treated them with the V-ATPase inhibitor bafilomycin. This led to tau accumulation in the AD-derived, but not the control-derived neurons. The data suggest that neurons in AD might be more susceptible to subtle changes in lysosomal pH balance.
What about Aβ?
In Stutzmann’s induced neurons, blocking the V-ATPase left Aβ unaffected. That’s not to say this peptide is insulated from changes in vesicular trafficking. Scientists know that amyloidogenic processing of amyloid precursor protein (APP) takes place on late endosomes. This process is enhanced by genetic variants, for example loss-of-function mutations in SORL1 (Aug 2019 news; Jun 2018 news). SORL1 encodes the protein SORLA, part of a cellular-protein-sorting conglomerate called the retromer. Retromers traffic APP away from late endosomes and to the Golgi, protecting it from cleavage by γ-secretase. Alas, in LA, Jessica Young, University of Washington, Seattle, reported that things are far more complex than that, revealing new pathways by which SORLA can shunt APP.
Like Karch, Young also uses human iPSCs and CRISPR editing. Her lab has knocked out or overexpressed SORL1, and tested how induced neurons would respond. At AAIC, Allison Knupp from Young’s lab reported that SORL1 KO cells had more APP in early endosomes and less in the trans Golgi, as would be expected. But there was also less APP in Rab9+ endosomes that recycle to the Golgi independently of the retromer. In keeping with this, when colleague Swati Mishra overexpressed SORL1, she found more APP in Rab9+ endosomes cycling to the Golgi, and also more APP in Rab11+ endosomes that recycle to the cell surface. “The data suggest there are more ways to less amyloidosis than through the canonical retromer,” Young told Alzforum. Indeed, she reported that SORL1 knockout and overexpression increased and decreased, respectively, production of Aβ1-40 and Aβ1-42 in induced neurons.
Karch and Young acknowledge that reprogramming cells to make iPSCs erases the epigenetic modifications cells have accrued during aging and thereby rejuvenate the stem cells relative to their fibroblast “ancestors.” For this reason, Young also experiments with direct conversion. This entails transmogrifying fibroblasts directly into neurons, bypassing the pluripotent stem cell step (Jun 2011 news; Jan 2013 news). Researchers led by Fred Gage at the Salk Institute, La Jolla, California, had previously reported that this process may preserve the transcriptomic age of the donor cells (Oct 2015 news).
Young, in collaboration with Dirk Keene and Suman Jayadev, both at UWashington, is testing this approach. They are starting out with fibroblast-like cells from the leptomeninges of autopsy-confirmed AD patients. They will run both iPSC and direct-conversion protocols side by side on the same tissue sample, then characterize the derived neurons to determine which best models Alzheimer’s pathology. Eventually, Young wants to use patient-derived neurons to test how a person’s genetic background influences endosomal trafficking, and defects thereof.
“It’s a huge collaborative effort,” Young said. “We plan to determine single-nucleotide polymorphism burden. We’ll analyze all the SNPS in endosome-related genes, group them by high and low risk, then see how they correlate with defects in trafficking, ultimately relating the phenotypes back to the genetics.” Young thinks this could identify very early changes in AD pathology that are not detected at autopsy. “You may have 100 people with a diagnosis of AD, but how that came about and progressed at the cellular level may be very different. If you subdivide based on genetics and cell biology, then you might be able to target treatments accordingly,” she said. “This is where derived neurons are really useful.”—Tom Fagan
References
Mutations Citations
Research Models Citations
News Citations
- Familial AD Mutations, β-CTF, Spell Trouble for Endosomes
- Gaining Notoriety, SORL1 Claims Spot Among Top Alzheimer’s Genes
- Turning Human Fibroblasts Into Neurons; Making Safer Stem Cells
- Stem Cells: Simpler to Make, Easier on the Immune System
- Neurons Derived Directly from Skin Cells Act the Age of Their Donors
Paper Citations
- Jiang S, Wen N, Li Z, Dube U, Del Aguila J, Budde J, Martinez R, Hsu S, Fernandez MV, Cairns NJ, Dominantly Inherited Alzheimer Network (DIAN), International FTD-Genomics Consortium (IFGC), Harari O, Cruchaga C, Karch CM. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry. 2018 Dec 13;8(1):265. PubMed.
- Yap CC, Digilio L, McMahon LP, Garcia AD, Winckler B. Degradation of dendritic cargos requires Rab7-dependent transport to somatic lysosomes. J Cell Biol. 2018 Sep 3;217(9):3141-3159. Epub 2018 Jun 15 PubMed.
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.