In Big Picture, Familial AD’s Biomarker Data Resemble LOAD
Quick Links
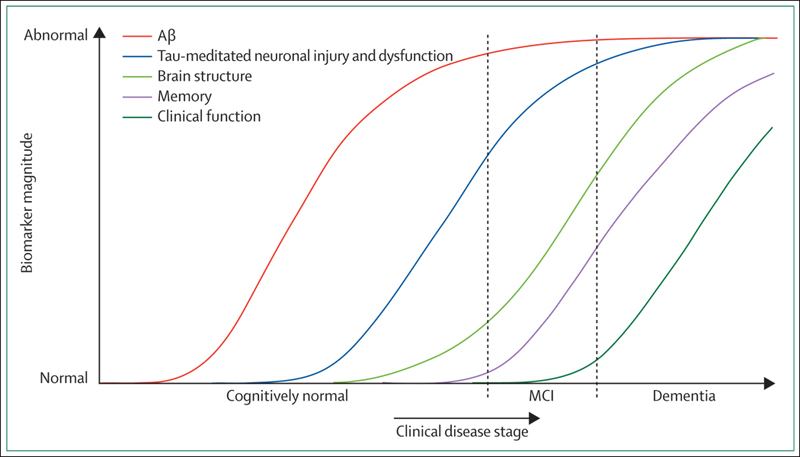
Researchers with eyes peeled toward prodromal Alzheimer’s disease probably see this diagram in their sleep—the one where the field’s five most validated markers trace their poignant path from normal cognition to fully developed AD. Two research groups proposed this theoretical model some years ago (see Perrin et al., 2009; Jack et al., 2010). But is it true? At least in its broad strokes, the answer seems to be yes, according to early data pouring in from biomarker studies in autosomal-dominant AD. By and large, brain amyloid, brain metabolism, atrophy, and functional connectivity data from independent analyses of the Dominantly Inherited Alzheimer Network (DIAN) and the Alzheimer’s Prevention Initiative (API) jibe with each other and confirm the sequence of preclinical biomarker changes proposed for late-onset AD. This article highlights DIAN and API neuroimaging results reported 14-19 July 2012 at the Alzheimer’s Association International Conference (AAIC) in Vancouver, Canada.
{kind=link}
DIAN and API came onto the scene in the last few years as scientists decided to act on the desperate need and untapped research potential in people with deterministic mutations that cause early onset familial AD (see ARF DIAN series and ARF API series for background on these initiatives). These folks comprise barely one percent of AD cases, but they develop the disease at younger ages and with predictable timing. This makes them ideal for charting biomarker changes from asymptomatic stages to full-blown dementia. Early onset AD patients are eager to participate in research; to date, trials of most experimental drugs have excluded them. On that, the tide is starting to turn. The U.S. National Institutes of Health announced in May that it would help fund the first-ever AD prevention trial in at-risk adults with no outward signs of AD—namely, Colombian people with the “Paisa” (E280A) presenilin 1 mutation who are enrolled in the API registry. Slated to start in 2013, the $100 million trial will test the monoclonal antibody crenezumab (see ARF related news story; see also ARF news story). DIAN—the study of the globally scattered families whose Alzheimer’s stems from a mutation in the amyloid precursor protein (APP) or presenilin (PS) genes—is also gearing up for therapeutic trials (see ARF related news story). So is another effort called Anti-Amyloid Treatment for Asymptomatic AD or A4, which intends to enroll healthy seniors with amyloid-positive brain scans (see AAIC story; ARF Q&A).
In Vancouver, Randall Bateman of Washington University School of Medicine, St. Louis, Missouri, recapped DIAN findings reported recently in the New England Journal of Medicine (Bateman et al., 2012) and in talks and posters throughout AAIC. At the AAIC’s imaging consortium pre-meeting, WashU’s Tammie Benzinger and colleagues won the Best Poster award for their analysis of brain amyloid, glucose metabolism, and cortical atrophy in 191 DIAN participants, including 119 mutation carriers. This is a more recent, larger version of the cohort of 128 analyzed for the NEJM report. The carriers had detectable brain amyloid some 15 years in advance of symptoms, metabolic decline and hippocampal shrinkage about 10 years prior, followed by cortical thinning and disrupted functional connectivity about five years before signs of disease. “These findings were reported for DIAN across three labs and two continents, using different analytic and statistical approaches to the data,” wrote Benzinger, who leads the DIAN imaging core, in an e-mail to Alzforum. At present, these data constitute cross-sectional comparisons; longitudinal data are still being gathered.
While the pattern of the DIAN imaging data for the most part matches what scientists see in sporadic AD, it is not identical. Unexpectedly, the DIAN cohort showed presence of white matter disease in mutation carriers. This is common in people with sporadic AD and is usually attributed to comorbid conditions such as high blood pressure or diabetes. However, DIAN participants are younger and generally do not have these comorbidities. “More investigation is needed to see whether the white matter findings are a consequence of the grey matter amyloid plaques or are perhaps related to amyloid deposition in blood vessels in the DIAN cohort,” Benzinger said. An expected difference on amyloid PET imaging is early deposition in basal ganglia, which has been reported previously in some cases of autosomal-dominant but not in sporadic AD.
In a featured research session on neuroimaging for presymptomatic trials, Alex Becker of Massachusetts General Hospital, Boston, presented initial data on baseline glucose metabolism in the brains of DIAN participants. Becker’s team analyzed how metabolism differed with mutation status and age (i.e. years from expected symptom onset) in 124 people who were either cognitively normal with a Clinical Dementia Rating (CDR) score of 0, or mildly symptomatic with CDR 0.5 or 1. As expected, symptomatic mutation carriers had lower brain activity—measured with fluorodeoxyglucose (FDG) positron emission tomography (PET)—than the control group of cognitively normal non-carriers. Moreover, Becker reported, the FDG signal showed a similar regional pattern as in sporadic AD, with metabolism waning in the precuneus and other AD-vulnerable brain areas, and falling off more steeply as carriers became more impaired. When comparing only asymptomatic subjects, mutation carriers had greater metabolic decline than non-carriers in two areas—an AD-vulnerable aggregate region and the angular gyrus. The hypometabolism became more pronounced as carriers approached their family’s age of symptom onset.
Furthermore, the researchers examined functional connectivity in the brains of DIAN participants. With this imaging modality, too, they saw differences between asymptomatic mutation carriers and non-carriers. Jasmeer Chhatwal of MGH and colleagues looked at resting-state connectivity in the default-mode network in functional MRI scans of 120 DIAN participants (83 mutation carriers and 37 non-carriers from the same families). People with sporadic AD are known to have decreased connectivity in their default mode network—a group of brain areas that fire up when the mind is resting (ARF related news story on Greicius et al., 2004). Similarly, mutation carriers in the DIAN cohort had decreased connectivity throughout the default-mode network, compared to non-carriers, even if they were still asymptomatic with a CDR of 0. Also like sporadic AD, functional connectivity in DIAN subjects decreased more quickly as they approached their family’s age of symptom onset, and continued to worsen with disease progression (see image below). “We are very excited about this data,” Chhatwal told the AAIC audience. Intriguingly, it looked as though the youngest subjects actually had increased connectivity, but this needs further study with larger sample sizes, he said. At last year’s AAIC in Paris, scientists at the Banner Alzheimer’s Institute, Phoenix, Arizona, reported that among 18- to 26-year-olds enrolled in API, mutation carriers already show differences in fMRI patterns (see ARF related news story), prior to amyloid deposition measurable by PIB. In the future, Chhatwal and colleagues plan to look at functional connectivity in other brain networks besides default mode, and to analyze whether the fMRI data correlate with amyloid imaging, CSF Aβ and non-amyloid biomarkers, as well as with specific mutation types.
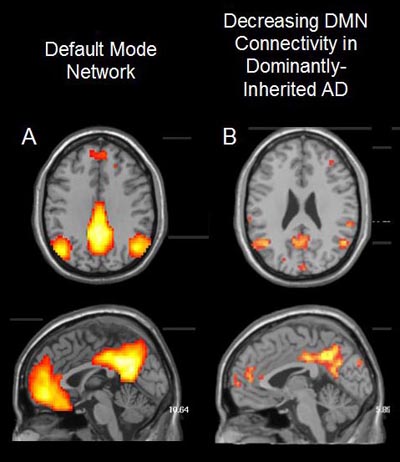
Adults with autosomal dominant Alzheimer’s disease mutations show decreased connectivity throughout the default-mode network (DMN) relative to non-mutation carriers, as revealed by functional magnetic resonance imaging (fMRI) brain scans. Image courtesy of Jasmeer Chhatwal, Massachusetts General Hospital
Reisa Sperling of Brigham and Women’s Hospital, Boston, is a DIAN site leader who has seen the functional MRI data from both DIAN and API cohorts. “There is remarkable convergence in the evidence that the default-mode network is dysfunctional prior to any cognitive impairment, and perhaps even prior to amyloid deposition in some subjects.” Sperling wrote in an email to Alzforum.
Now on to API data. At the Vancouver meeting, Adam Fleisher of the Banner Alzheimer’s Institute in Phoenix, Arizona, spoke about florbetapir amyloid PET, cerebrospinal fluid (CSF), and MRI biomarker changes in API participants at preclinical and clinical stages of early onset AD. Fleisher had presented some of the florbetapir data in January at the Human Amyloid Imaging Conference in Miami, Florida (see ARF related news story). On the whole, biomarker profiles in API mutation carriers looked similar to the hypothesized trajectories for sporadic AD, Fleisher wrote to Alzforum. Fibrillar Aβ deposits cropped up about 16 years before clinical symptoms, followed a few years later by CSF Aβ42 and subsequently tau changes, and then hippocampal shrinkage about seven years in advance of symptoms.
Given that adult carriers as young as 18 to 26 already appeared to have fMRI and even some subtle brain structure changes compared with non-carriers, the API scientists reached back even further in age and decided to look for changes in 9- to 17-year-old children with the Paisa PS1 mutation. Yakeel Quiroz of Boston University, Massachusetts, and Universidad de Antioquia, Colombia, and colleagues scoured MRI scans of 21 mutation carriers in this youth bracket. They measured cortical thinning in nine signature regions previously defined in patients with mild sporadic AD (Dickerson et al., 2009). This MRI signature can predict which MCI patients will convert to AD (Bakkour et al., 2009) and, in a longitudinal study, predicted which healthy seniors eventually develop AD (Dickerson et al., 2011). Even in this youth bracket, mutation carriers were distinguishable, but in a surprising way—by having a thicker cortex than age-matched non-carriers. However, the young carriers lost brain tissue more quickly and had a thinner cortex, on average, than age-matched non-carriers by the time they reached their 30s. Older, symptomatic carriers continued to show faster cortical thinning in all nine signature regions, as well as in the signature’s summary measure.
Among the AD prevention initiatives, API has the most genetically homogeneous participants, as all have the same PS1 mutation. DIAN participants have one of dozens of familial AD mutations, and the A4 cohort is heterogeneous, enrolling any asymptomatic senior with biomarker evidence of amyloid. MRI profiles may, however, differ according to FAD mutation. David Cash and Kirsi Kinnunen at University College London, U.K., analyzed structural MRI data from a local cohort of presymptomatic FAD mutation carriers, some of whom also enrolled in DIAN. Thirty-one people with PS1 mutations and 12 with APP mutations received several MRI scans about a year apart and were followed longitudinally for up to two decades. The analysis included 44 age- and sex-matched non-carriers, each scanned twice about 18 months apart. By the time symptoms appeared, mutation carriers already had higher atrophy rates than non-carriers in all structures analyzed (whole brain, ventricles, hippocampus); however, the MRI profiles were not the same in all carriers. APP mutation carriers seemed to lose tissue faster in the hippocampus, while PS1 carriers showed accelerated atrophy in the global measures of whole brain shrinkage and ventricular expansion, Cash reported on a poster.
Likewise, structural MRI profiles differed between PS1 and APP carriers in a cross-sectional analysis of 190 DIAN participants, as presented on a poster by Kinnunen. This cohort comprised 119 mutation carriers (95 PS1, 10 PS2, 14 APP) and 71 non-carriers. When the PS1 mutation carriers were studied separately, presymptomatic carriers had smaller whole-brain volumes than non-carriers. However, this finding disappeared when brain volumes from all mutation carriers were lumped together and compared with non-carriers, suggesting that individual FAD mutations may affect brain volume differently. What do these tidbits of data on different imaging modalities, age brackets, and mutations amount to at this point? “I think the data coming out on DIAN and API biomarkers looks overall great so far. The bottom line is that things seem consistent with the amyloid cascade,” noted WashU’s David Holtzman, who sits on the DIAN steering committee. “It looks very encouraging in the sense that there is quite a long window where one could initiate prevention trials.” Nick Fox of the London group agreed, but added that in terms of designing those trials, the early biomarker changes in asymptomatic FAD mutation carriers present both a challenge as well as an opportunity. “Do we aim for trials in individuals many years before symptom onset, perhaps at a stage when there is very little evidence of any irreversible changes? Or do we aim for subjects who are closer to onset age, when tracking progression and assessing effects of therapy would be easier and quicker?” he wrote to Alzforum.—Esther Landhuis.
References
News Citations
- DIAN: Registry for eFAD to Chart Alzheimer’s Preclinical Decade
- Colombians Come to Fore in Alzheimer’s Research, Mass Media
- NIH Director Announces $100M Prevention Trial of Genentech Antibody
- A Close Look at Passive Immunotherapy Newbie, Crenezumab
- DIAN Grows, Gets Ready for Therapeutic Trials
- Collaborative Umbrella CAPs Three Prevention Trial Initiatives
- Anti-Amyloid Treatment in Asymptomatic AD Trial
- Network Diagnostics: "Default-Mode" Brain Areas Identify Early AD
- Reeling In Biomarker Data in Young Carriers, API Rocks Staging Boat
- Miami: When Does Amyloid Deposition Start in Familial Alzheimer’s?
Paper Citations
- Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature. 2009 Oct 15;461(7266):916-22. PubMed.
- Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010 Jan;9(1):119-28. PubMed.
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
- Greicius MD, Srivastava G, Reiss AL, Menon V. Default-mode network activity distinguishes Alzheimer's disease from healthy aging: evidence from functional MRI. Proc Natl Acad Sci U S A. 2004 Mar 30;101(13):4637-42. PubMed.
- Dickerson BC, Bakkour A, Salat DH, Feczko E, Pacheco J, Greve DN, Grodstein F, Wright CI, Blacker D, Rosas HD, Sperling RA, Atri A, Growdon JH, Hyman BT, Morris JC, Fischl B, Buckner RL. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex. 2009 Mar;19(3):497-510. PubMed.
- Bakkour A, Morris JC, Dickerson BC. The cortical signature of prodromal AD: regional thinning predicts mild AD dementia. Neurology. 2009 Mar 24;72(12):1048-55. PubMed.
- Dickerson BC, Stoub TR, Shah RC, Sperling RA, Killiany RJ, Albert MS, Hyman BT, Blacker D, Detoledo-Morrell L. Alzheimer-signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology. 2011 Apr 19;76(16):1395-402. PubMed.
Other Citations
External Citations
Further Reading
Papers
- Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012 Aug 30;367(9):795-804. PubMed.
News
- Network Diagnostics: "Default-Mode" Brain Areas Identify Early AD
- Anti-Amyloid Treatment in Asymptomatic AD Trial
- NIH Director Announces $100M Prevention Trial of Genentech Antibody
- A Close Look at Passive Immunotherapy Newbie, Crenezumab
- Colombians Come to Fore in Alzheimer’s Research, Mass Media
- Miami: When Does Amyloid Deposition Start in Familial Alzheimer’s?
- Experimental α7 Agonist Meets Cognitive and Clinical Endpoints
- Q&A With Roche’s CNS Leader Luca Santarelli
- When Is a C9ORF72 Repeat Expansion Not a C9ORF72 Repeat Expansion?
- Wave of New BACE Inhibitors Heading to Phase 2
- Researchers Join to Draw Posterior Cortical Atrophy Out of Shadows
- CSF Markers: Goodbye, Research Use Only; Hello, Clinical
- Cochrane Asks for Field’s Input on Draft Reporting Standards
- Collaborative Umbrella CAPs Three Prevention Trial Initiatives
- DIAN: Registry for eFAD to Chart Alzheimer’s Preclinical Decade
- Reeling In Biomarker Data in Young Carriers, API Rocks Staging Boat
- DIAN Dispatch from Hawaii: Glimpse at Data, Push for Trials
- DIAN Forms Pharma Consortium, Submits Treatment Trial Grant
- DIAN Grows, Gets Ready for Therapeutic Trials
- New Assays for Aβ Oligomers in CSF Claim Femtogram Sensitivity
- Metrology, Certification Heavies Take CSF Tests Under Their Wings
- Can We All Agree on How to Draw a Hippo(campus)?
- On Risk, Benefit, and More, Prevention Initiatives Take Questions
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.