Mutations
MAPT G389R (G>C)
Quick Links
Overview
Pathogenicity: Frontotemporal Dementia : Pathogenic
Clinical
Phenotype: Frontotemporal Dementia
Reference Assembly: GRCh37/hg19
Position: Chr17:44101376 G>C
dbSNP ID: rs63750512
Coding/Non-Coding: Coding
DNA
Change: Substitution
Expected RNA
Consequence: Substitution
Expected Protein
Consequence: Missense
Codon
Change: GGG to CGG
Reference
Isoform: Tau Isoform Tau-F (441 aa)
Genomic
Region: Exon 13
Findings
This mutation was first identified in a 38-year-old patient who presented with progressive aphasia and memory disturbance (Murrell et al., 1999; Ghetti et al., 2000). Apathy followed, along with other symptoms including hyperphagia, rigidity, pyramidal signs, and ultimately a profound dementia. Death occurred at age 43. A paternal uncle had also died at age 43 with similar symptoms. Autopsy showed numerous Pick body-like inclusions, see below.
This mutation was also identified in a case of corticobasal syndrome (CBS) (Rossi et al., 2008). The patient was a 41-year-old man with progressive asymmetric signs of cortical and basal ganglia involvement consistent with CBS. MRI showed asymmetric cortical atrophy. Family history was negative for similar illnesses. The mutation was present in the patient's unaffected elderly father, suggesting possible incomplete penetrance.
Most recently, the G389R mutation was identified in a young Frenchwoman with juvenile onset frontotemporal dementia (Chaunu et al., 2013). The proband was just 17 years old when she developed changes in mood, attention, and memory. She was initially diagnosed with depression, but her condition worsened, and she developed pronounced cognitive difficulties, language impairment, and social disinhibition. She developed extrapyramidal features such as distal tremor, rigidity, and akinesia. Her eye movements were slow and she developed vertical gaze palsy. She became mute, unable to walk, and developed seizures. She died seven years after symptom onset. Her paternal uncle had died at age 37, following a rapidly progressive dementia. Likewise, her paternal grandfather had died at age 46 with a rapidly progressing dementia and was thought to have had Pick’s disease. Her great-great-uncle had died at age 42, mute and with mood disorders and a left pyramidal syndrome. The patient's paternal great-grandmother died at age 73 without dementia, despite the fact that she likely carried the mutation. The patient’s father carried the mutation but was asymptomatic at age 46 except for very mild and stable difficulties in executive functioning. Overall, the family history suggests autosomal dominant transmission of the G389R MAPT mutation, but with variable penetrance.
Neuropathology
One proband's brain showed numerous tau-positive Pick body-like inclusions in the neocortex and numerous tau-positive filamentous inclusions in axons in the frontal, temporal, and parietal lobes, and severe cerebral atrophy (Murrell et al., 1999; Ghetti et al., 2000).
Autopsy of the young French patient showed severe atrophy in the frontal and temporal lobes, including the primary motor areas. Only the occipital pole was largely spared. The ventricles were dilated and the corpus callosum very thin. The basal ganglia were normal with the exception of mild atrophy in the caudate. The substantia nigra and locus coeruleus were pale. The main lesions were neuronal loss, astrocytosis, and laminar supragranular spongiosis, especially in the most atrophic cortical areas. White matter was diffusely demyelinated. Severe neuronal loss was observed in the substantia nigra as well. Tau immunohistochemistry revealed rounded neuronal inclusions mimicking Pick bodies. Some neurons also had diffuse cytoplasmic tau staining. Affected neurons were largely restricted to cortical layers 2 and 3. Numerous tau-positive threads and coiled bodies were observed in the subcortical white matter, putamen, and brainstem. A few spherical lesions were also present in the dentate gyrus. No abnormalities were detected using antibodies directed against Aβ, TDP-43, neurofilament, FUS, or α-synuclein (Chaunu et al., 2013).
Biological Effect
Recombinant G389R tau showed a reduced ability to promote microtubule assembly. The mutation also weakened tau binding to microtubules, and resulted in a greater aggregation propensity of tau isoforms, including those containing three-repeat (3R) domains and those with four-repeat (4R) domains.
Last Updated: 09 Dec 2016
References
Paper Citations
- Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999 Dec;58(12):1207-26. PubMed.
- Ghetti B, Murrell JR, Zolo P, Spillantini MG, Goedert M. Progress in hereditary tauopathies: a mutation in the Tau gene (G389R) causes a Pick disease-like syndrome. Ann N Y Acad Sci. 2000;920:52-62. PubMed.
- Rossi G, Marelli C, Farina L, Laurà M, Maria Basile A, Ciano C, Tagliavini F, Pareyson D. The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov Disord. 2008 Apr 30;23(6):892-5. PubMed.
- Chaunu MP, Deramecourt V, Buée-Scherrer V, Le Ber I, Brice A, Ehrle N, El Hachimi K, Pluot M, Maurage CA, Bakchine S, Buée L. Juvenile Frontotemporal Dementia with Parkinsonism Associated with Tau Mutation G389R. J Alzheimers Dis. 2013 Jan 1;37(4):769-76. PubMed.
Further Reading
Papers
- Gemignani A, Pietrini P, Murrell JR, Glazier BS, Zolo P, Guazzelli M, Ghetti B. Slow wave and rem sleep mechanisms are differently altered in hereditary pick disease associated with the TAU G389R mutation. Arch Ital Biol. 2005 Feb;143(1):65-79. PubMed.
- Goedert M, Satumtira S, Jakes R, Smith MJ, Kamibayashi C, White CL 3rd, Sontag E. Reduced binding of protein phosphatase 2A to tau protein with frontotemporal dementia and parkinsonism linked to chromosome 17 mutations. J Neurochem. 2000 Nov;75(5):2155-62. PubMed.
Learn More
Protein Diagram
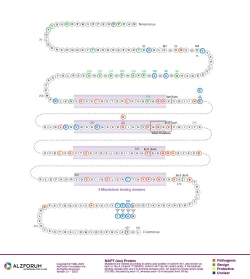
Primary Papers
- Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M. Tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol. 1999 Dec;58(12):1207-26. PubMed.
- Ghetti B, Murrell JR, Zolo P, Spillantini MG, Goedert M. Progress in hereditary tauopathies: a mutation in the Tau gene (G389R) causes a Pick disease-like syndrome. Ann N Y Acad Sci. 2000;920:52-62. PubMed.
- Rossi G, Marelli C, Farina L, Laurà M, Maria Basile A, Ciano C, Tagliavini F, Pareyson D. The G389R mutation in the MAPT gene presenting as sporadic corticobasal syndrome. Mov Disord. 2008 Apr 30;23(6):892-5. PubMed.
Other mutations at this position
Alzpedia
Disclaimer: Alzforum does not provide medical advice. The Content is for informational, educational, research and reference purposes only and is not intended to substitute for professional medical advice, diagnosis or treatment. Always seek advice from a qualified physician or health care professional about any medical concern, and do not disregard professional medical advice because of anything you may read on Alzforum.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.