Microglial Regulation and Function Scrutinized at Heidelberg Meeting
Quick Links
New insights into microglial biology and function created quite the buzz at Mechanisms of Neurodegeneration, an EMBO/EMBL symposium held June 14-17. Organized by Karin Dumstrei, Todd Golde, Christian Haass, and Bart De Strooper, the second iteration of this meeting drew about 200 researchers to the EMBO training and conference center in Heidelberg, Germany. Topics ran the gamut from old and knotty questions such as how γ-secretase works, to hot topics like microglial pruning of synapses, to completely unexpected ideas, i.e., can microglia seed plaques?
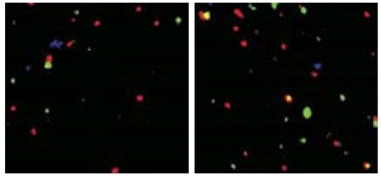
Saving Synapses.
Hippocampal CA3 tissue from APPPS1 mice (left) has less presynaptic Vglut2 (green) and postsynaptic GluR1 (red) than CA3 in APPPS1 C3 knockouts (right). [Courtesy of Shi et al., 2017; Science Translational Medicine/AAAS.]
Beth Stevens, Children’s Hospital, Boston, set the tone for the meeting by reviewing the role of microglia in Alzheimer’s and Huntington’s diseases. Over the last few years, seminal work has changed how the field views microglia in neurodegeneration. Scientists discovered how these immune cells resume a job they once had early in brain development and then retired; that is, to prune synapses. Central to this lies the complement signaling cascade, kick-started by complement C1q and fueled by complement C3, which flags structures that are to be engulfed by phagocytic cells. Stevens outlined recently published data from her and Cynthia Lemere’s labs at Brigham and Women’s Hospital, in which knocking out C3 protected APP/PS1 mice from learning and memory loss, albeit at the cost of much more Aβ than usually accrues in this AD model (see Jun 2017 news).
In her talk in Heidelberg, Stevens extended this concept to Huntington’s disease. She reported that C1q and C3 are upregulated in mice expressing mutant human huntingtin with an expanded polyglutamine repeat, and that the animals retain more synapses after chronic treatment with an anti-C1q antibody. “That they find the exact same thing going on in parallel in two different models strongly suggests that we have a common mechanism for synaptic pruning in neurodegenerative diseases,” said Haass, from the German Center for Neurodegenerative Diseases (DZNE) in Munich. “For me, this was one of the highlights of the meeting and other people seemed to think so, as well,” Haass told Alzforum. Stevens said she is now testing whether complement deficiency affects huntingtin pathology and motor function in HD mice.
Stevens’ work supports the idea that phagocytic microglia simultaneously clear amyloid plaques and prune synapses. In contrast to that, might these cells also contribute to plaque formation? Michael Heneka from the DZNE in Bonn showed data on protein conglomerates called ASC specks. Short for “apoptosis-associated speck-like protein containing a caspase-recruitment domain,” ASC aggregates when activated by the inflammasome, forming bundles that appear as specks under the light microscope. ASC specks activate caspase-1, which in turn activates inflammatory cytokines such as interleukin 1β and IL-18, powering an inflammatory cascade. Released by microglia, ASC specks can be taken up by nearby cells, unleashing the same inflammatory cascade in them (Jun 2014 news). Heneka previously reported that microglia in the frontal cortices of AD patients spew out ASC specks. In keeping with this, he found that silencing the NLRP3 inflammasome that activates ASC can protect APP/PS1 mice from learning and memory deficits (Dec 2012 news).
What then, causes the release of ASC specks in AD? In Heidelberg, Heneka said he initially suspected Aβ, but experiments done in his lab to prove this hunch did not pan out. That’s because it turned out that ASC specks have a voracious affinity for binding Aβ. While that appetite complicated the experiments, it led Heneka to a more important idea—that ASC specks help seed and spread Aβ deposits in the brain. With a movie of microglia under the microscope, Heneka showed that Aβ latches onto ASC specks like iron filings to a magnet. He went on to describe, in a series of in vitro biochemical and cell biology experiments, which regions of the ASC structure bind Aβ, how ASC can pull Aβ out of solution by gravity, and that ASC specks seed formation of Aβ oligomers.
Does this matter in vivo? Heneka reported that ASC always turns up in Aβ plaques in mouse models of AD, and crossing those models with ASC knockouts dramatically reduces levels of soluble and insoluble Aβ in APP/PS1 mice, while also rescuing their learning and memory deficits. Haass called this work a hot new topic. “That Michael finds the core of Aβ plaques always have ASC specks is super surprising,” Haass said.
All told, Heneka’s data suggest that ASC specks could play a role in the seeding and spreading of plaques. He tested this by injecting Aβ-containing lysates into mouse brains as done in now-classic Aβ seeding experiments by Mathias Jucker and Lary Walker (see, for example, Sep 2006 news on Meyer-Luehmann et al., 2006). When Heneka injected three-month-old APP/PS1 mice with Aβ it increased the amount of deposits around the brain five months later, as seen on serial brain sections. However, in APP/PS1 x ASC knockouts, injected lysates caused no ramp-up in plaque deposition. ELISA assays of total brain Aβ backed this up.
Haass considered the data compelling. “I have found that Aβ injected into the brain has never really seeded well—you have to flood the brain with peptide. So there is something missing in regular seeds [that’s needed for full propagation], and the ASC speck may be it,” he told Alzforum.
What about ASC specks in patients? Preliminary evidence suggests they play a role there, too. Heneka and colleagues have immunoprecipitated the complex from samples of postmortem brain tissue; in the case of AD and mild cognitive impairment, this pulls down Aβ as well. They detected no Aβ in pulldowns from normal age-matched controls or from people who had had frontotemporal dementia or corticobasal degeneration. In fact, Heneka showed an image of a brain section from an AD patient that displayed what he called “microglial Armageddon,” with an abundance of Iba1-expressing microglia littered with adjacent ASC specks. Side by side with early drawings of microgliosis penned by Alois Alzheimer himself, the two looked remarkably similar.
Scientists at the meeting avidly discussed Heneka’s presentation. Marco Colonna, Washington University, St. Louis, asked if microglia digest the ASC speck/Aβ complexes—au contraire, Heneka said they are extremely stable and resist degradation. Stevens wondered if astrocytes play a role in this ASC pathology—Heneka plans to investigate that. He did note that astrocytes appear to atrophy more at sites of ASC release, but since Aβ pathology is rampant there, too, Heneka can’t be sure which might be driving astrocyte cell death.
Others questioned the role of ASC specks in AD pathology. Konrad Beyreuther from the University of Heidelberg was perplexed that ASC specks reportedly have higher affinity for Aβ40 than for Aβ42. “Why then do we only see Aβ42 in human plaques?” he asked. Heneka had no immediate answer for that, but others speculated that Aβ42 might be needed to template misfolding and aggregation of more Aβ on the ASC speck core.
Other scientists described new findings on the homeostasis of microglia. Researchers led by Frank Heppner at Charité – Universitätsmedizin Berlin had previously reported that myeloid cells infiltrating the brain were strangely uninterested in mopping up amyloid plaques (Apr 2015 conference news and Prokop et al., 2015). Why might that be? In Heidelberg, Heppner posited that something in the AD brain suppresses the cells. He reported that peripheral myeloid cells that infiltrate the brain in normal mice sport more dynamic soma and cellular processes than do myeloid cells infiltrating the brain of an AD mouse model. Researchers in his lab found that an interleukin cascade might be silencing the cells in the disease state. Knocking down p40, a subunit of both IL12 and IL23, reduced plaque pathology in APP/PS1 transgenic mice (Nov 2012 news). Heppner said that p40 and IL12 levels climb in AD patients, while a polymorphism in the IL23 receptor has been linked to AD in Han Chinese (Hu et al., 2012; Sudduth et al., 2012; Liu et al., 2014). Heppner now has data to suggest that the cognate receptors for IL12/23 in the brain lie on astrocytes, suggesting that a cytokine-mediated cross-talk between these cells and microglia temper the latter. To test that, researchers in his lab have conditionally knocked out the IL12 and IL23 receptors in astrocytes and crossed those animals with APP/PS1 mice. Time will tell whether the offspring are also protected from amyloidosis like the IL12/23 knockouts.
Heppner’s data might not only shed light on microglial deficiencies in AD, but could have implications for therapy as well. He reported that Aβ immunotherapy is ineffective in AD mouse models whose microglia are depleted; indeed, the more microglia there are, the more efficacious the antibodies. Heppner also reported a proof of the peripheral sink hypothesis, which posits that mopping up Aβ in the blood speeds up its clearance from the brain. He generated mice that make IgM antibodies against Aβ in the periphery and showed that they have more Aβ in the blood but one-third less plaque burden than controls. Haass called these experiments important. “This data fit nicely with our report that TREM2 deficiency reduces antibody-induced clearance of amyloid,” he said. “This confirms that we really need microglia for immunotherapy to work.”
This meeting featured a smidgeon of new data about TREM2, when Colonna confirmed and extended findings from the Haass lab that this microglial receptor plays major roles in microglial homeostasis and reported the new finding that TREM2 functions in the regulation of autophagy, as well. Evidence for the latter came from examining TREM2-negative microglia in 5xFAD mice. The cells were packed with multilamellar vesicles, suggestive of increased autophagic clearance, and they had accumulated lipidated LC3, an autophagosomal marker. In keeping with this, Colonna reported weaker signaling through mammalian target of rapamycin (mTOR) in TREM2-negative microglia. A central regulator of autophagy, mTOR coordinates growth and nutrient signals in cells. Colonna found that TREM2-negative microglia were unable to muster normal levels of many of the intermediaries generated by classic metabolic pathways. These include glycolysis, the pentose phosphate shunt, and the tricarboxylic acid cycle.
All told, Colonna concluded that the absence of TREM2 signaling suppresses normal metabolic function in microglia, which then try to compensate by increasing autophagy. “Temporarily, that counterbalance might be good, but eventually the cells become apoptotic and die,” he said.—Tom Fagan
References
Research Models Citations
News Citations
- Sans Complement: Amyloid Grows, Synapses and Memory Stay
- Inflammasomes Spread from Cell to Cell
- Microglia and AD—Does the Inflammasome Drive Aβ Pathology?
- Double Paper Alert—A Function for BACE, a Basis for Amyloid
- Could Adaptive Immunity Set the Brakes on Amyloid?
- Soothing Neuroinflammation Quells Plaques in Mice
Paper Citations
- Meyer-Luehmann M, Coomaraswamy J, Bolmont T, Kaeser S, Schaefer C, Kilger E, Neuenschwander A, Abramowski D, Frey P, Jaton AL, Vigouret JM, Paganetti P, Walsh DM, Mathews PM, Ghiso J, Staufenbiel M, Walker LC, Jucker M. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science. 2006 Sep 22;313(5794):1781-4. PubMed.
- Prokop S, Miller KR, Drost N, Handrick S, Mathur V, Luo J, Wegner A, Wyss-Coray T, Heppner FL. Impact of peripheral myeloid cells on amyloid-β pathology in Alzheimer's disease-like mice. J Exp Med. 2015 Oct 19;212(11):1811-8. Epub 2015 Oct 12 PubMed.
- Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, Grossman M, Xiong C, Craig-Schapiro R, Clark CM, Pickering E, Kuhn M, Chen Y, Van Deerlin VM, McCluskey L, Elman L, Karlawish J, Chen-Plotkin A, Hurtig HI, Siderowf A, Swenson F, Lee VM, Morris JC, Trojanowski JQ, Soares H, . Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012 Aug 28;79(9):897-905. PubMed.
- Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer's disease. Neurobiol Aging. 2012 Oct 9; PubMed.
- Liu Y, Yu JT, Zhang W, Zong Y, Lu RC, Zhou J, Tan L. Interleukin-23 receptor polymorphisms are associated with Alzheimer's disease in Han Chinese. J Neuroimmunol. 2014 Jun 15;271(1-2):43-8. Epub 2014 Mar 21 PubMed.
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.