Alzheimer’s Risk Genes Give Up Some Secrets at SfN
Quick Links
Large genome-wide association studies have brought the number of Alzheimer’s disease risk genes to more than 20, but how do those common genetic variants modify risk? That the basic function of many of these genes remains shrouded in mystery has not helped scientists answer this question, but at this year’s Society for Neuroscience meeting, held October 17-21 in Chicago, a little more of the veil started to slip. Much as researchers initially expected that these 20 genes would throw the field open wide beyond the amyloid hypothesis, thus far, it appears that some GWAS genes help regulate the two hallmarks of AD: plaques and tangles.
Many labs have used knockout approaches to study GWAS hits, a caveat since most of the common polymorphisms that associate with AD weakly modulate gene transcription and alter risk by as little as 10 percent. Nevertheless, data from knockouts may be informative. As Steve Estus, University of Kentucky, Lexington, pointed out, just because GWAS SNPs have weak effects doesn’t mean we shouldn't target those genes for therapeutic purposes. “If we can pharmacologically modulate those genes by, say, 80 percent, then we may achieve a significant clinical effect,” he said.
Case in point, the gene for clusterin (aka Apolipoprotein J). Researchers in John Fryer’s lab at the Mayo Clinic, Jacksonville, Florida, crossed Clu knockouts with APP/PS1 mice. Graduate student Aleksandra Wojtas reported that the homozygote knockouts shifted Aβ deposition from the parenchyma to the blood vessels, where it deposited to cause cerebral amyloid angiopathy. Other researchers were struck by the stark contrast between knockouts and controls, some calling it the cleanest data they’d seen at the meeting (see image below).
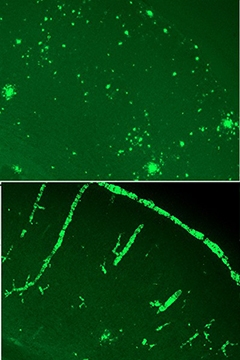
Cluless.
Without clusterin, APP/PS1 mice accumulate Aβ predominantly in the blood vessels (bottom). [Image courtesy of John Fryer.]
“We were pretty surprised,” said Fryer. “We expected perhaps fewer plaques, but to have everything showing up as CAA was unexpected.” When Fryer was a graduate student in David Holtzman’s lab at Washington University, St. Louis, Ron DeMattos had made a similar cross with PDAPP mice and saw no strong redistribution of Aβ to the vasculature (see Jul 2002 conference news). What explains the difference between the models? Fryer told Alzforum that in general, the PDAPP mice have very little CAA, but CAA begins to emerge when the PDAPP/Clu-negative mice age. Fryer has also crossed Clu knockouts with a mouse model of CAA, and vessel pathology runs rampant then as well. “We think this clusterin CAA effect is real,” Fryer said.
Holtzman thought the findings fit with the role of apolipoproteins in the brain. “It is consistent with the fact that clusterin, like ApoE, influences Aβ aggregation and seeding,” he wrote to Alzforum. “Where ApoE promotes CAA, clusterin somehow inhibits its formation.”
What explains Aβ’s dramatic shift to the blood vessels? Fryer suggested two possibilities. The lack of Clu might render the blood vessels stickier, allowing them to trap Aβ. However, since knocking out Clu in a wild-type background caused no obvious change in blood vessels or behavioral phenotype, Fryer favors the second possibility: that without clusterin, ApoE, the other major lipoprotein in the brain, funnels Aβ through the interstitial fluid to the blood vessels, where it gets trapped (see model below).
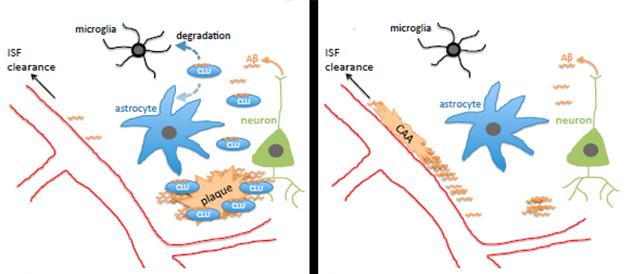
Wojtas further reported that the Clu knockouts had fewer dystrophic neurites in the brain than APP/PS1 controls. Fryer expected this, since neurites generally become dystrophic around parenchymal plaques. Less expected, however, was that astrogliosis and microgliosis in the knockouts paled in comparison with those in the APP/PS1 brain, and the knockouts spewed less cytokines, suggesting little or no inflammatory response. “We only saw robust gliosis when a few plaques formed, otherwise there seemed to be no inflammation in the knockouts despite a tremendous amount of CAA,” said Fryer.
How do these findings shed light on the association between genetic Clu variants and AD? The single nucleotide polymorphism found in GWAS protects against AD and also seems to increase expression of clusterin. That might suggest that it favors sequestration of Aβ in plaques rather than in the vasculature, but it might also promote degradation of Aβ by microglia (see model above). “We need to look at the APP/PS1 Clu heterozygotes, which more realistically model the human situation where clusterin levels are altered but not absent,” said Fryer. Holtzman noted that human clusterin may act differently to the mouse homolog as well. “That will be important to assess,” he said. Fryer plans to generate humanized CLU mice and develop in vitro assays to see if clusterin alters fibril formation of both Aβ40 and Aβ42, including peptides containing the Dutch and Iowa mutations, which lead to CAA.
Researchers believe that another GWAS hit, TREM2, might also boost microglial clearance of Aβ. However, researchers from Bruce Lamb’s lab at the Cleveland Clinic, Ohio, reported at last year’s SfN meeting that knocking out the TREM2 gene appeared to have little effect on amyloid plaques in the cortices of APP/PS1 mice and even lowered plaque burden in their hippocampi (see Dec 2014 conference news). TREM2 took the field by storm when GWAS identified variants in the gene that almost tripled the risk for AD, making this the strongest genetic risk factor for the disease after ApoE4 (see Nov 2012 news). If this microglial receptor is not required for Aβ clearance, then what about the other major hallmark of AD, neurofibrillary tangles? In Chicago, Shane Bemiller from Lamb’s lab reported that TREM2 helps clear phosphorylated and aggregated forms of tau from mouse brain.
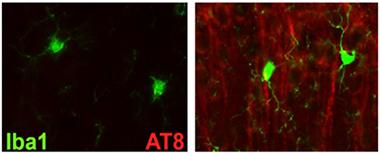
Tau Trouble.
Tau immunoreactivity ramps up in the cortices of htau mice that lack TREM2 (right). [Image courtesy of Shane Bemiller.]
Bemiller crossed TREM2 knockouts with htau mice, which overexpress human tau in place of the endogenous mouse version. These mice express all six isoforms of the human protein and accumulate phosphorylated tau and neurofibrillary tangles as they age. Bemiller reported a huge increase in AT8 and AT180 binding in the cortices of TREM2-negative htau mice compared with htau controls. AT8 and AT180 antibodies recognize various phosphorylated forms of tau and paired helical filaments, precursors to neurofibrillary tangles. AT8 reactivity was mostly in the superficial layers of the cortex, whereas AT180 binding encroached into deeper layers, said Bemiller. In the hippocampus, only AT8 binding surpassed that in control htau mice. However, Gallyas staining revealed that these mice accumulated more NFTs in both the cortex and hippocampus than did controls.
Bemiller concluded that knocking out TREM2 compromises microglial clearance of tau. Interestingly, he found an uptick in the microglial activation marker Iba1 in the vicinity of AT8 immunoreactivity, suggesting that the glia do respond to the tau aggregates but fail to clear them effectively. Other scientists asked if the Iba1 may be on some other cells instead of microglia. Bemiller conceded the possibility, but pointed to the difficulty of identifying peripheral monocytes that enter the brain. Lamb told Alzforum that data from sorted cells and bone marrow chimeras suggests that microglia mediated the effects on tau pathology.
Other evidence suggests that the tau problems in these TREM2 knockouts run deeper than clearance. Phosphorylation of kinases such as JNK, ERK1/2, and GSK3BETA crept up in these mice compared to htau controls, suggesting that hyperphosphorylation of tau might be out of control. Phospho-JNK, normally found in the nucleus, appeared mostly in the perinuclear space in neurons. Bemiller suggested that lack of TREM2 compromises cross-talk between glia and neurons, disrupting cell signaling pathways and causing dysregulation of various tau kinases.
Steve Estus’ lab focused on a different GWAS hit that might also be involved in Aβ clearance. The ATP-binding cassette transporter ABCA7 is found on microglia and may regulate phagocytosis. GWAS uncovered a single nucleotide polymorphism, rs3764650T, which protects against AD (see Apr 2011 news). Estus found that this single nucleotide polymorphism (SNP) increases expression of the protein (Vasquez et al., 2013).
Last March, researchers at deCODE Genetics in Iceland reported a SNP (rs200538373G>C) in intron 41 of the gene that increases risk for AD. That second SNP seems to muck up splicing, causing an extension to exon 41 that introduces a premature stop codon. Scientists at deCODE predicted that rearrangement leads to loss of function (see Mar 2015 news). Estus has looked for the exon extension in tissue samples taken from AD patients and controls. In Chicago, Jared Vasquez from his lab reported that he was indeed able to amplify both the normal and the extended exon forms of ABCA7 from RNA taken from a donor who carried a C allele at that SNP. Surprisingly, Vasquez also found the extended exon in two of six samples taken from people homozygous for the more common G allele. Estus said that some other SNP in the gene may modulate splicing of that exon, though he told Alzforum they have not been able to find a variant that fits the bill. Alternatively, some dietary or lifestyle factors might modulate ABCA7 splicing, said Estus. Vasquez is examining medical records to see if these donors share any history in common that might explain the ABCA7 expression pattern.
Several SNPs near the complement receptor 1 (CR1) gene also associate with late-onset AD (see Jul 2009 conference news). The receptor plays a role in the innate immune system, where it limits complement-mediated damage to host cells. Because it also promotes phagocytosis, researchers believe it may contribute to Aβ clearance. However, interpreting GWAS data has been mired in the biological complexity surrounding this receptor. Red blood cells and peripheral phagocytes make several isoforms, and soluble forms circulate in the plasma. Scientist know less about CR1 in the central nervous system.
Andrea Tenner of the University of California, Irvine, has localized forms of CR1 by immunohistochemistry. In Chicago, she reported that two monoclonal antibodies, 8C9.1 and J3B11, bind CR1 in human brain, and immunoreactivity seems specific to astrocytes. Other labs have detected CR1 on neurons, but Tenner claimed this may be non-specific. In her hands, pre-absorption of antibody solutions with CR1 abolished binding to astrocytes, but not neurons. Neurons may carry non-CR1 epitopes to which the antibodies bind, she suggested. She said it is important for the field that antibody reactivity be carefully characterized.
Using these antibodies, Tenner found that while the extent of CR1 antibody binding varied among different brain samples, no correlation emerged with AD diagnosis or with genotype of the GWAS SNPs, rs6656401 and rs4844609. This suggests that the AD risk alleles do not alter expression of CR1 in the brain. Tenner reported that the risk alleles do slightly increase the affinity of CR1 for their known ligands, the complement proteins C1q and C3b. However, she does not believe this explains the disease association either, because tighter binding would predict better clearance of complement-bound Aβ, which would likely be protective.
One possible explanation, Tenner said, could be poor clearance through the plasma due to lower expression of CR1 on red blood cells. GWAS risk alleles correlate tightly with reduced expression of a long form of CR1 on erythrocytes (see Mahmoudi et al., 2015). In support of this, Tenner noted that less Aβ binds red blood cells in AD patients than in age-matched controls, and that people tend to lose CR1 from erythrocytes as they age (Rogers et al., 2006; Moldenhauer et al., 1988). These observations point to an important role for erythrocyte CR1 in Aβ clearance, but Tenner also noted controversy around the peripheral sink hypothesis. Others at the meeting were intrigued by the data, particularly the finding that only a subset of astrocytes seemed to produce CR1. Some wondered if astrocytes might phagocytose Aβ and how that might be regulated, and if astrocyte expression of CR1 changes with age.
Overall, from the study of GWAS hits thus far, a sense emerged that some of the genes are involved in either clearance of toxic peptides, or their production (see Part 6 of this series).—Tom Fagan
References
Alzpedia Citations
Research Models Citations
News Citations
- Stockholm: Clusterin (or Beware the Evil Chaperone!)
- TREM2 Data Surprise at SfN Annual Meeting
- Enter the New Alzheimer’s Gene: TREM2 Variant Triples Risk
- Large Genetic Analysis Pays Off With New AD Risk Genes
- Massive Icelandic Genome Analysis Offers Clues to Health and Disease, Including AD
- Vienna: New Genes, Anyone? ICAD Saves Best for Last
- Alzheimer’s GWAS Hits Point to Endosomes, Synapses
Paper Citations
- Vasquez JB, Fardo DW, Estus S. ABCA7 expression is associated with Alzheimer's disease polymorphism and disease status. Neurosci Lett. 2013 Nov 27;556:58-62. Epub 2013 Oct 18 PubMed.
- Mahmoudi R, Kisserli A, Novella JL, Donvito B, Dramé M, Réveil B, Duret V, Jolly D, Pham BN, Cohen JH. Alzheimer's disease is associated with low density of the long CR1 isoform. Neurobiol Aging. 2015 Apr;36(4):1766.e5-12. Epub 2015 Jan 9 PubMed.
- Rogers J, Li R, Mastroeni D, Grover A, Leonard B, Ahern G, Cao P, Kolody H, Vedders L, Kolb WP, Sabbagh M. Peripheral clearance of amyloid beta peptide by complement C3-dependent adherence to erythrocytes. Neurobiol Aging. 2006 Dec;27(12):1733-9. Epub 2005 Nov 14 PubMed.
- Moldenhauer F, Botto M, Walport MJ. The rate of loss of CR1 from ageing erythrocytes in vivo in normal subjects and SLE patients: no correlation with structural or numerical polymorphisms. Clin Exp Immunol. 1988 Apr;72(1):74-8. PubMed.
Other Citations
Further Reading
No Available Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
RIKEN Center for Brain Science
Please let me make the following comments on John Freyer's work, because this is very important for the research of the entire AD community to go in the right direction.
A small concern about this paper, and others that use similar mice, is that the transgenic mouse model of AD used overexpresses mutant APP and PS. Overexpression of membrane proteins containing 1 and 9 transmembrane domains, respectively, could induce non-specific ER stress. Overexpression of APP and PS transgenes results in the destruction of at least two gene loci in the host animals and may, in principle, generate artificial phenotypes.
It is surprising that none of the transgenic mice, to my knowledge, have been sequenced for defects. APP interacts with kinesin via JIP-1, hence APP overexpression may perturb axonal transport, causing axonal sprouting. In addition, APP overexpression results in overproduction of non-Aβ APP fragments. In particular, CTF-β—which does not accumulate much in human AD brain and is recognized by most anti-Aβ antibodies—is known to be more toxic than Aβ.
Based on these concerns, I would suggest that the authors examine whether their main findings are the result of APP and PS overexpression and not innate disease processes. We are willing to provide free to academic scientists our model mice that overproduce Aβ42 without overexpressing APP or PS1 (Saito et al., 2014). Right now, more than 130 laboratories all over the world are using these mice. We estimate that approximately 60 percent of the phenotypes in conventional APP Tg mice are artifacts. I invite the authors and other scientists experimentally working on AD in transgenic mouse models to recognize this and take steps to correct the APP overexpression paradigm.
References:
Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer's disease. Nat Neurosci. 2014 May;17(5):661-3. Epub 2014 Apr 13 PubMed.
Make a Comment
To make a comment you must login or register.