APP Upp: Mutation Nixes Six Amino Acids from Aβ, Spurs Aggregation
Quick Links
Thirty missense mutations in amyloid precursor protein are known to cause autosomal-dominant Alzheimer’s disease. Now, scientists at Uppsala University, Sweden, have identified a deletion in the APP gene that does the same thing. At the 14th International Conference on Alzheimer’s and Parkinson’s Diseases, held March 27–31 in Lisbon, Portugal, María Pagnon de la Vega described a mutation that leads to the loss of six amino acids from the mid-region of the Aβ peptide. It segregated with disease in a single family.
- A pathogenic APP deletion results in shortened Aβ34 and Aβ36 peptides.
- Aβ36 aggregates faster than wild-type or mutant Aβ42.
- Plaques contain mostly Aβ36 and are unusually round.
Cleavage of this mutant APP by BACE1 and then γ-secretase produces truncated peptides, Aβ34 and Aβ36. Intriguingly, the shortened Aβ36 peptide appears particularly prone to aggregation. “To our knowledge, this is the first deletion in APP found to cause autosomal-dominant AD,” Pagnon de la Vega said in Lisbon.
Working with Lars Lannfelt, Dag Sehlin, and Martin Ingelsson at Uppsala, Pagnon de la Vega identified the “Uppsala” deletion in a family whose affected members developed symptomatic AD at about 40 years of age. The researchers traced its inheritance through three generations, where the disease followed an autosomal-dominant pattern. Their study focused on two afflicted brothers, age 43 and 45, one of whom later died. The brothers had an affected parent who died of AD. They had typical tau tangles by neuropathology and elevated cerebrospinal fluid tau markers, as well as the expected brain atrophy and hypometabolism in temporal and parietal cortex.
Notably, however, their CSF Aβ, as measured by standard assays, resembled that of healthy controls. Ingelsson told Alzforum that the antibodies used in CSF assays also detect Aβ34 and Aβ36, as their epitopes are not in the deleted region. In contrast, PiB PET scans were mildly positive, with an SUVR of about 1.4. In the brother who came to autopsy, his cortical amyloid plaques contained mostly Aβ36 or smaller fragments and looked unusually round, with sharply defined edges. The researchers found almost no Aβ40 or Aβ42 in these deposits. They also found plaques in the occipital cortex, where few tend to form in most cases of sporadic AD.
When the researchers examined the behavior of synthetic Aβ34 and Aβ36 in vitro, they found that Aβ36 fibrillizes rapidly, far faster than wild-type Aβ42 or even Aβ containing the Arctic mutation.
Pagnon de la Vega noted that the pattern of aggregation is different as well. The researchers were unable to detect protofibrils, the intermediate stage of aggregation, by ELISA and electron microscopy; instead, fibrils popped up right away. The Swedish researchers have made a double transgenic UppSwe mouse; it accumulates mostly Aβ36 as well. The researchers are currently making antibodies against the shortened peptides.
To further characterize the new mutation, Pagnon de la Vega and colleagues transfected cell lines with Uppsala APP. They found high levels of Aβ34 in the medium, but no sign of the sAPPα cleavage fragment by Western blot. The deletion may shift α-secretase cleavage toward the N-terminus, so that the resulting fragment is no longer recognized by standard antibodies, Pagnon de la Vega surmised. She is currently analyzing the media by mass spectrometry to find out if α-secretase cleavage occurs.
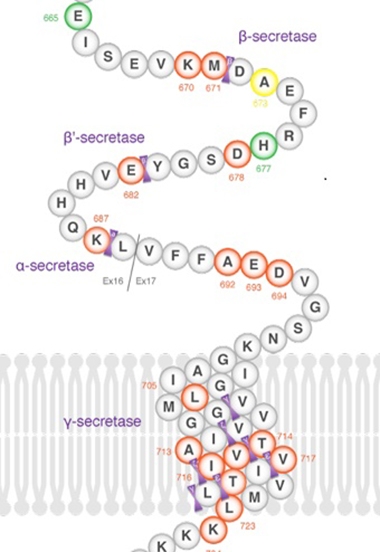
Deletion, Where Art Thou? A newly discovered APP mutation leads to Aβ peptides that are missing six amino acids. Researchers at AD/PD were coy about which ones they are.
The audience in Lisbon was much intrigued by the findings, and expressed some dismay when the scientists would not disclose the location of the deletion.
The same group, led by Lannfelt, has identified previous APP mutations. The Swedish mutation alters two amino acids next to the BACE1 cleavage site and stimulates this cut, boosting production of Aβ40 and Aβ42 (Mullan et al., 1992). The Arctic mutation results in Aβ that readily forms protofibrils. People with this mutation develop AD early, but have few amyloid cored plaques and are negative by PiB PET. Arctic protofibrils were used to develop the BAN2401 antibody that has entered Phase 3 trials.
At the same location as the Arctic mutation, a single amino acid deletion has been linked to AD in three Japanese families (Tomiyama et al., 2008). This Osaka mutation appears recessive, and homozygous carriers have an unusual form of AD, with few plaques by PiB PET, and often develop motor problems and difficulty walking. This mutant Aβ readily forms oligomers, but no fibrils.
APP also comes in at least one protective variant; carriers of the Icelandic mutation produce about 20 percent less Aβ over their lifespan.—Madolyn Bowman Rogers
References
Mutation Interactive Images Citations
Mutations Citations
Therapeutics Citations
Paper Citations
- Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992 Aug;1(5):345-7. PubMed.
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008 Mar;63(3):377-87. PubMed.
Further Reading
Annotate
To make an annotation you must Login or Register.
Comments
No Available Comments
Make a Comment
To make a comment you must login or register.